As
filed with the Securities and Exchange Commission on
Registration No. 333-
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form
REGISTRATION STATEMENT
UNDER
THE SECURITIES ACT OF 1933
| (Exact Name of Registrant as Specified in its Charter) |
| 3845 | ||||
(State or other jurisdiction of incorporation or organization) |
(Primary Standard Industrial Classification Code Number) |
(I.R.S. Employer Identification No.) |
+
(Address, including zip code, and telephone number, including area code, of principal executive offices)
(Address, including zip code, and telephone number, including area code, of agent for service)
Copies to:
Oded Har-Even Ron Ben-Bassat Sullivan and Worcester LLP 1633 Broadway, New York, NY 10019 Tel: (212) 660-3000 |
Barry I. Grossman, Esq. Matthew Bernstein, Esq. Ellenoff Grossman & Schole LLP 1345 Avenue of the Americas, 11th Floor New York, NY 10105 Tel: (212) 370-1300 |
Approximate date of proposed sale to public: As soon as practicable on or after the effective date of this registration statement.
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933 check the following box. ☒
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See definitions of “large accelerated filer,” “accelerated filer,” and “smaller reporting company”, and “emerging growth company” in Rule 12b-2 of the Exchange Act. (Check one):
| Large accelerated filer ☐ | Accelerated filer ☐ |
| Smaller
reporting company | |
| Emerging
growth company |
If
an emerging growth company, indicate by checkmark if the registrant has elected not to use the extended transition period for complying
with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act.
The Registrant hereby amends this Registration Statement on such date or dates as may be necessary to delay its effective date until the Registrant shall file a further amendment which specifically states that this Registration Statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act, as amended, or until this Registration Statement shall become effective on such date as the Commission, acting pursuant to such Section 8(a), may determine.
The information in this prospectus is not complete and may be changed. We may not sell these securities until the Securities and Exchange Commission (“SEC”) declares our registration statement effective. This prospectus is not an offer to sell these securities and is not soliciting an offer to buy these securities in any state where the offer or sale is not permitted.
SUBJECT TO COMPLETION, DATED ______, 2024
Preliminary Prospectus
IR-Med, Inc.
_______ Units
Each Unit Consisting of
One Share of Common Stock
and
One Warrant to Purchase One Share of Common Stock
This is a firm commitment public offering of _____ Units at an assumed offering price of $ per unit, each Unit consisting of one share of common stock, par value $0.001 per share, and one warrant to purchase one share of Common Stock (the “Units”) of IR-Med, Inc. Each warrant is immediately exercisable for one share of common stock at an exercise price of $[_] per share (or 100% of the price of each share of common stock sold in the offering) and will expire [_] years from the date of issuance. The Units will not be certificated and the shares of common stock and the warrants comprising such Units are immediately separable and will be issued separately in this offering.
Prior to this offering, there has been a limited public market for our common stock on the OTCQB® Market, or OTCQB. On June 21, 2024, the last reported sale price of our common stock as reported on the OTCQB was $0.60 per share. We intend to apply to list our common stock and warrants on the Nasdaq Capital Market under the symbols “IRME” and “IRMEW”, respectively. No assurance can be given that our application will be approved. If our application is not approved or we otherwise determine that we will not be able to secure the listing of our common stock on Nasdaq, we will not complete this offering.
We have assumed a public offering price of $ , which represents the last reported sales price of our common stock as reported on the OTCQB on , 2024. There is no assurance that this offering will be completed, or as to the terms of this offering. In addition, the closing sales price of our common stock as reported on the OTCQB may not be indicative of the final offering price or market price of our common stock on Nasdaq and there can be no assurance that a trading market will develop for our shares of common stock on Nasdaq. The final public offering price will be determined through negotiation between us and the Underwriter in the offering and the recent market price used throughout this prospectus may not be indicative of the final offering price. In addition, quotes of stock trading prices on an over-the-counter marketplace may not be indicative of the market price on a national securities exchange.
Investing in our common stock involves a high degree of risk. Please read “Risk Factors” beginning on page 21 of this prospectus.
Neither the SEC nor any state securities commission has approved or disapproved of these securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
| Per Unit | Total | |||||||
| Public offering price | $ | $ | ||||||
| Underwriting discounts and commissions (1) | $ | $ | ||||||
| Proceeds, before expenses, to us | $ | $ | ||||||
(1) We have agreed to give the Underwriter a discount equal to eight percent (8%) of the gross proceeds of this offering. We refer you to “Underwriting” for additional information regarding Underwriter compensation.
We have granted the underwriters a 45-day option to purchase up to additional shares of common stock and/or warrants.
Delivery of the Units is expected to be made on or about , 2024.
Sole Book-Running Manager
Maxim Group LLC
The date of this prospectus is ____, 2024
TABLE OF CONTENTS
| 2 |
Neither we nor the Underwriter have authorized anyone to provide any information or to make any representations other than those contained in this prospectus or in any free writing prospectus prepared by or on behalf of us or to which we have referred you. We and the Underwriter take no responsibility for and can provide no assurance as to the reliability of, any other information that others may give you. This prospectus is an offer to sell only the shares offered hereby, but only under circumstances and in jurisdictions where it is lawful to do so. The information contained in this prospectus or in any applicable free writing prospectus is current only as of its date, regardless of its time of delivery or any sale of shares of our common stock. Our business, financial condition, results of operations and prospects may have changed since that date.
Neither we nor the Underwriter have done anything that would permit this offering or possession or distribution of this prospectus or any free writing prospectus we may provide to you in connection with this offering in any jurisdiction where action for that purpose is required, other than in the United States. You are required to inform yourselves about and to observe any restrictions relating to this offering and the distribution of this prospectus and any such free writing prospectus outside of the United States.
| 3 |
PROSPECTUS SUMMARY
This summary highlights information contained elsewhere in this prospectus. This summary is not complete and does not contain all of the information you should consider in making your investment decision. Before investing in our common stock, you should carefully read this entire prospectus, especially the sections titled “Risk Factors” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and our financial statements and the related notes included elsewhere in this prospectus. Unless the context otherwise requires, the terms “IR-Med,” “the Company,” “we,” “us,” “our” and similar references in this prospectus refer to IR-Med, Inc. a Nevada corporation.
Overview
We were incorporated in the State of Nevada in April 2007 under the name “Monster Motors, Inc.” We began operating the business of IR. Med Ltd., an Israeli company, through a reverse acquisition on December 24, 2020. IR. Med Ltd. (an Israeli company which was founded in 2013) continues to operate as our operating subsidiary, and we are the sole stockholder of IR. Med Ltd. Our corporate headquarters and research facilities are located at ZHR Industrial Zone, Rosh Pina, Israel.
We are in the process of developing point-of-care decision support devices based on the patented cutting-edge infrared spectroscopy and artificial intelligence (AI) analysis technology platform, as a basis for point-of-care decision support devices. The electrooptic visual and infrared spectroscopy technology platform allows harmless and non-invasive gathering of bio-information from a patient’s blood and tissue. Bioinformation is then analyzed using our AI-based algorithms to provide healthcare professionals with decision support in the detection and monitoring of various disease conditions. We plan to use the proceeds from this offering to continue development efforts of our products, while mainly focusing on the DiaSafe device, production of commercial units, marketing, and working capital.
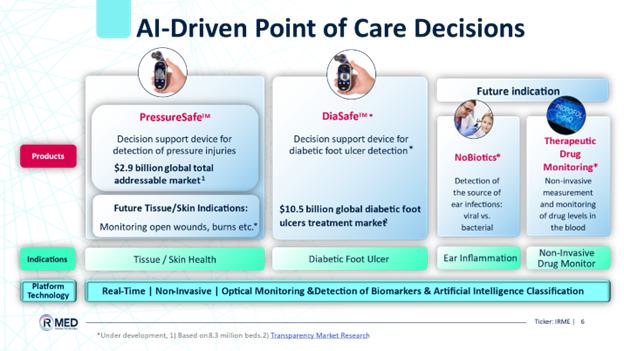
PressureSafe: Our first product based on this platform, is a handheld device designed to revolutionize the early detection of pressure injuries (“PIs”) affecting the skin and underlying tissue. PIs in the U.S. alone account for $26.8 billion in healthcare spending and result in 60,000 deaths annually. PressureSafe is expected to contribute to early detection of PIs, regardless of patient skin tone. This will drive equitable healthcare and help reduce the toll and cost of PIs. We plan to launch PressureSafe as a decision support system (DSS) tool for caregivers in hospitals, nursing homes, and home-care companies. On April 9, 2024, the PressureSafe decision support device received a U.S. Food and Drug Administration (FDA) listing certification. PressureSafe is classified as a Class I device. We are currently working on completing the development of the commercial version of the PressureSafe device, planned to be launched during September 2024, following the listing under the FDA.
DiaSafe: Similarities in the physiological development of PIs and diabetic foot ulcers (“DFU”) under the skin surface allow the IRMED PressureSafe device to be adopted to support the early detection of DFU among diabetic patients at high risk of developing DFU. We are assessing and planning the development of our second product, which is a handheld optical monitoring device that will support early detection of DFUs in lower limb skin and underlying tissue, primarily caused by prolonged pressure on the sole among diabetic patients a condition, which sometimes is accompanied by other comorbidities as lower limb neuropathy.
Our novel technology platform will enable direct assessment of the development of a DFU before it becomes an open wound that may lead to limb amputation. The Israeli Innovation Authority, or IIA, has approved our plan to develop a diabetic foot ulcer device for early detection of DFU. On January 25, 2024, the Israel Innovation Authority (the “IIA”) approved a program to develop a device for the early detection of diabetic foot ulcers among diabetic patients, with a project budget of NIS 3,761,978 (approximately US$ 1,030,000) which includes an amount equal to 50% grant of the total budget provided at the time of the grant, disbursed in installments over the course of 13 months, by the project’s progress. In consideration for the grant by the IIA, the subsidiary is required to pay royalties at the rate of 3%-5% from the total sales until the repayment date of the full amount of the grant, plus annual interest at the SOFR rate. In addition, the IIA must approve any arrangement whereby the Company seeks to transfer the technology relating to the project, or its development, from Israel. Following the IIA grant we plan to commence a clinical trial in the center of Israel’s leading diabetes clinic.
| 4 |
Future indication as part of our research and development is an innovative otoscope, Nobiotics, to support physicians with an immediate indication as to whether mid-ear infection (otitis media), a common malady in children, is of a bacterial origin and thus requiring antibiotic treatment, or of a viral origin that consequently does not require antibiotic treatment.
Our technology platform utilizes AI. AI is a broad term generally used to describe conditions where a machine mimics “cognitive” functions associated with human intelligence, such as “learning” and “problem-solving.” Basic AI includes machine learning, where a machine uses algorithms to parse data, learn from it, and then suggest a determination or prediction about a given phenomenon. The machine is “trained” using large amounts of data and algorithms that provide it with the ability to learn how to perform various tasks.
The global diagnostics market is driven in large by solutions that can be applied in healthcare settings, as these tools will drive decisions regarding specific treatments and the associated outlays. However, despite advances in medical imaging and other diagnostic tools, misdiagnosis remains a common occurrence.
Our initial focus is on the development of decision support system solutions utilizing our proprietary platform for the pre-emptive diagnosis of PIs, and diabetic foot ulcers. Our current business plan focuses on two principal medical devices:
| 1. | PressureSafe, a handheld skin-agnostic optical monitoring device that is being developed to support early detection of PIs to the skin and underlying tissue, primarily caused by prolonged pressure associated with bed confinement; and | |
| 2. | DiaSafe, a handheld optical monitoring device that is being developed to support early detection of DFUs in lower limb skin and underlying tissue, primarily caused by prolonged pressure on the sole among diabetic patients a condition, which sometimes is accompanied by other comorbidities as lower limb neuropathy. |
| 5 |
Fig1. IRMED AI-Driven Point of Care Decisions technology platform
Overview of Target Market and Our Solutions
Pressure Injury Market
Populations are aging due to improvements in healthcare. However, there are increased rates of obesity, diabetes and cardiovascular diseases. This combination of an increasingly aging population and such diseases has resulted in more people with decreased mobility needing assistance with activities of daily living. A major morbidity of decreased mobility is development of PIs. PIs develop as a result of a combination of physiologic events and external conditions. Along with localized, oedema, ischemia and reperfusion hindering injury to tissues, impaired lymphatic drainage and mechanical deformation of tissue cells have been shown to contribute to pressure injury.
Compression prevents lymph fluid drainage and leads to deterioration in tissue cell normal activities, which causes increased interstitial fluid and waste build up, contributing to the development of PIs. The time required to develop PIs depends on many factors, including the patient’s physiological medical background and the degree of pressure and sheer force placed on the tissue. PIs occur over predictable pressure points where bony protuberances are more likely to compress tissues when the patient is in prolonged contact with hard surfaces. Studies show that the heel area is the second most frequent location for a pressure ulcer, with the most prevalent being the sacrum. The heel accounts for between 23% and 28% of all pressure ulcers.1
While the overall number of Hospital Acquired Conditions (HAC) have decreased by 8%, pressure injuries have resisted improvement efforts and continue to grow by 10% annually. PIs are both costly and deadly. The U.S. Agency for Healthcare Research and Quality (AHRQ) reports that PIs add $10.2 billion to annual U.S. healthcare costs. Furthermore, these are associated with over 45% of the 63,619 HAC related deaths in the U.S., making it the leading HAC related death.2

(AHRQ, 2019). Source: https://www.ahrq.gov/data/infographics/hac-rates_2019.html; AHRQ National Scorecard on Hospital-Acquired Conditions Final Results for 2014-2017 (PDF, 787 KB).
PIs impose a tremendous healthcare burden. As stated in the National Pressure Injury Advisory Panel fact sheet for 2023, 60,000 patients die every year as a direct result of pressure injuries. Acute care attributable to hospital-acquired PIs reaches $26.8 billion, and 2.5 million patients per year develop a PI. Patient care costs per PI range from $20,900 up to $151,700. PIs are among one of the five most common harms experienced by patients and the second most common claim for lawsuits, after wrongful death. More than 17,000 lawsuits arise due to PIs annually at an average settlement of $250,000. PIs occur across the healthcare industry, including in 10% of acute care patients, 25% of long-term acute care patients, 12% of nursing home patients and 12% of rehabilitation center patients.3
1 Smith, S., Ashby, S., Thomas, L. and Williams, F., 2017. Evaluation of a multifactorial approach to reduce the prevalence of pressure injuries in regional Australian acute inpatient care settings. International Wound Journal, 15(1), pp.95-105.
2 AP News. 2019. Pressure Ulcers Cost U.S. Healthcare $10.2 Billion and Contribute to Nearly 29,000 Hospital Deaths Each Year.
3 National Pressure Injury Advisory Panel fact sheet for 2023
| 6 |
The most common method used to detect early PIs is a visual assessment by a professional caregiver focusing on areas at high probability to develop PIs. This skin and tissue visual assessment is subjective, unreliable, untimely (as PIs often occur suddenly without visual cues), and only effective to detect PIs once they are visible. Technology-based methods for detecting and monitoring have been developed, but as far as we know, none have succeeded in providing an effective solution. Pressure injuries, especially HAPIs, are complex, difficult to treat, and at risk for re-occurrence.
Pressure Injuries Background
A pressure injury is caused when skin integrity is broken down by some type of unrelieved pressure, leading to the destruction of normal structure and function. The National Pressure Injury Advisory Panel (NPIAP), the preeminent U.S. professional organization dedicated to prevention and management of PIs, uses these four criteria to define a PI:
| ● | A pressure injury is localized damage to the skin and underlying soft tissue, usually over a bony prominence. | |
| ● | The injury can present as intact skin or an open ulcer and may be painful. | |
| ● | The injury occurs as a result of intense pressure, prolonged pressure, or pressure in combination with shear. | |
| ● | The tolerance of soft tissue for pressure and shear may also be affected by microclimate, nutrition, perfusion, comorbidities and condition of the soft tissue. |
Common places for PIs to develop include the back of the head, shoulders, elbows, buttocks, hips, ankles, and heels.
The 4 Stages of Pressure Injuries - PI Stage 1
Non-blanchable erythema of intact skin. Intact skin with a localized area of non-blanchable erythema, which may appear differently in darkly pigmented skin. Presence of blanchable erythema or changes in sensation, temperature or firmness may precede visual changes. Color changes do not include purple or maroon discoloration; these may indicate deep tissue pressure injury.
Stage 1 in Darkly Pigmented Skin: Research indicates that people with darker skin tones are more likely to develop higher stage pressure injuries, possibly because skin assessment protocols are less effective in identifying damage earlier. Pigmentation of the skin may prevent visualizing the reactive hyperemia in the pressure injury.4
Currently, PIs are discovered only as they begin to appear on the skin, after they have been festering underneath the skin layers. Nurses regularly assess patients at high risk by evaluating them according to accepted scores (e.g., Braden or Norton Scales). Hospitals can then get the patient onto a different type of mattress that wicks away moisture change patient support and reduces pressure and impose orders for the individual to be turned every few hours, for example. The risk of a PI among acute care patients ranges between 2-40% of patients
Intrinsic risk factors such as diabetes, malnutrition and smoking also increase the overall risk for pressure injuries. The spinal cord injury patient population is at the highest risk (25-66%) of developing a PI due to the combination of immobility and decreased sensation.] A prospective study of spinal cord patients not only found that sacral and ischial PIs were very common (43% and 15%, respectively), as might be expected, but also noted that the second most common location was on the heel (19%).5
Nursing home patients have PI prevalence of 11%6 and are most likely to develop PIs on the sacrum or heels. Nursing home patients were also found to have contractures at a prevalence of 55%. Contractures are caused by decreased elasticity of the tissue surrounding major joints, and the resulting lack of full mobility in the affected extremities significantly increases the risk of PI information.
4 Current Perspectives on Pressure Injuries in Persons with Dark Skin Tones from the National Pressure Injury Advisory Panel, Adv Skin Wound Care. 2023 Sep 1;36(9):470-480. doi: 10.1097/ASW.0000000000000032. Epub 2023 Aug 7. PMID: 37590446.
5 Delmore, B., Lebovits, S., Suggs, B., Rolnitzky, L. and Ayello, E., 2015. Risk Factors Associated with Heel Pressure Ulcers in Hospitalized Patients. Journal of Wound, Ostomy & Continence Nursing, 42(3), pp.242-248.
6 Palese, A., Zammattio, E., Zuttion, R., Ferrario, B., Ponta, S., Gonella, S. and Comoretto, R., 2020. Avoidable and Unavoidable Pressure Injuries Among Residents Living in Nursing Homes. Journal of Wound, Ostomy & Continence Nursing, 47(3), pp.230-235.
| 7 |
A significant market is the home healthcare market, which is anticipated to be worth $645 billion by 2025 (CAGR 8.7%).7 It is estimated that by 2030, seniors aged 65 and over will represent 20% of the U.S. population, and over 19 million seniors are estimated to need homecare services. Homecare companies have a strong incentive to prevent PIs as they are rated and carry part of the cost treating those patients.
According to a survey published in 2000 by UCLA School of Medicine,8 in a total sample of 3,048 patients, 9.12% had PIs, and of these, 37.4% had more than one PI, and 14% had three or more. Considering the worst PIs for each subject, 40.3% had Stage II and 27% had Stage III or IV injuries.
The Agency for Healthcare Research and Quality (AHRQ) has identified several basic principles for PI prevention: (a) use a validated tool to assess risk such as the Braden Scale and Norton Scale; (b) implement a preventive plan for residents at risk, which should focus on avoiding friction and sheer trauma to at-risk skin regions, as well as an individualized plan to reduce pressure, such as frequent repositioning; and (c) daily inspection of the skin for high-risk residents, as deep tissue damage can occur in as little as two hours. The most common method used to detect early pressure injuries is a visual assessment by a professional caregiver focusing on areas where PIs most frequently develop. This visual assessment is subjective, unreliable, untimely and ineffective as PIs develop under the skin before becoming visible to the naked eye. Technology-based methods for detecting and monitoring PIs have been developed, but none have succeeded in providing an effective solution. These include ulcer detection based on skin conductivity which has relatively low resolution and is influenced by different topical skin conditions (e.g., moisture, urine or feces). Other system solution methods such as electronic medical record programs, which prompt providers to document results of PI screening every shift or day, are of great importance in diagnosing PIs early and preventing progression. Pads designed to specifically cover pressure points such as the sacrum and heels, as well as foam pads designed to wrap around at-risk body parts, are common products. However, it is important to note that some pads can actually be detrimental; for example, supports with cut-outs can have increased pressure at their edges. Hospital-acquired PI rates are increasing while all other hospital-acquired conditions are decreasing (AHRQ, 2019).
PressureSafe
Over the past six years, we have been designing and developing PressureSafe, a novel device that has the potential to provide a reliable method of monitoring and recording patients, providing additional bio information to healthcare providers as to where and when a pressure injury may occur. The technology platform is designed to record information relating to each patient. The core technologies underlying the PressureSafe device are patent protected (US Patent No. US 10,709,365 and US Patent No. US10,772,541). Our technology platform is based on the fact that tissues of the human body absorb and reflect omitted light in different wave lengths (from the visual light to infra-red light), and the light is reflected and scattered back from different skin layers. During this process, the reflected and scattered light waves through a damaged area changes its properties in comparison to light reflection and scattering from normal healthy tissue. The PressureSafe device is being designed to capture, analyze and identify tissue status to make early PI diagnoses using Spectrographic Analysis, while AI based algorithm is implemented to improve diagnostic accuracy. The PressureSafe device illuminate the skin with a miniature set of LEDs less than a second in order to acquire the tissue fingerprint. The emitted light photons from the device will be absorbed, scattered, and reflected back. The device will then detect the absorption and reflectance, and by using algorithms, it will process the signals to identify and classify the scanned area.
As all person’s skin properties are unique, the diagnosing physician needs to use a device as the PressureSafe, which automatically calibrate the device to the specific patient’s skin, a process that takes merely a few seconds and allows personalized diagnosis, improving physician diagnostic process effectiveness, as the PressureSafe device is designed to measure regardless of skin color. Our technology is being developed to enable the assessment of different subepidermal layers by scanning through these skin layers, thus improving the identification of the damage, assessing the subepidermal damaged tissue volume and assisting with additional information to allow better treatment efficacy. Measuring the differences of subepidermal biomarkers has been developed to detect early formation of PIs and to “raise a flag” to allow the caregivers intervene and prevent their appearance. The biomarkers that our algorithm detects start from the early inflammatory process, as soon as local underlying tissue function is disturbed, and cells begin to be damaged.
7 Home Healthcare Market will grow at CAGR of 8.7% to hit $645.10 billion by 2025 : Adroit Market Research.
8 Ferrell, B., Josephson, K., Norvid, P. and Alcorn, H., 2000. Pressure Ulcers Among Patients Admitted to Home Care. Journal of the American Geriatrics Society, 48(9), pp.1042-1047.
| 8 |
PressureSafe is a hand-held scanner designed to provide additional information as a decision support system (DSS), to support the care giver effectively with the main diagnostic ability to identify PIs and to differentiate between deep tissue PIs (before they become visible) and Stage 1 PIs. Deep tissue PIs are serious, deep PIs that form under intact skin, spread in deep tissues and eventually present themselves as full thickness wounds. The PressureSafe is composed of (a) a handheld optic probe device, which utilizes harmless infra-red light that is placed on the skin and has a disposable tip that is changed between patients. The optic probe with its disposable cover is placed on suspected areas for performing measurements; (b) a disposable probe tip component, changed between patients to avoid cross-contamination; (c) a software component containing machine learning algorithm for analyzing the collected data; and (d) software for connectivity and downloading the collected data and measurements results to the EMR/EHR systems used by the medical center or homecare company.
PressureSafe is a non-invasive real-time optical monitoring device to support early intervention in PI treatment prior to skin breakage. The device performs a reflectance spectroscopy scan to generate information for the decision maker, while collecting data on epidermal and subepidermal physiological changes together with other bio-signals typical of early formation of PIs in the main three skin layers, thus detecting the appearance of life-risking pressure injuries. PressureSafe is designed to detect changes deep in the skin, regardless of skin tone, by measuring bio markers. As soon as local subcutaneous tissue function is disturbed and cells begin to disintegrate by pressure exerted upon the body area, our scanner is designed to be able to detect this very early inflammatory process and tissue structure changes. The technology will allow patient monitoring and immediate reading in a non-invasive way. It has the potential to help to reduce the number of PIs dramatically through accurate early classification, making it attractive for public and private healthcare systems worldwide.
PressureSafe Studies
Our product candidates are in various stages of development and production. The PressureSafe device is in an advanced stage of development and is planned to be our first go-to-market product.
We have completed the development of the first generation PressureSafe prototype in the second quarter of 2022. In June 2022, IR. Med Ltd., our wholly owned subsidiary, entered into a study agreement with Beit Rivka, a large geriatric hospital in Israel associated with Clalit, the largest Health Maintenance Organization (HMO) in Israel, to conduct a usability study of PressureSafe.
On July 17, 2023, we published our interim report of usability study performed in Israel in leading medical centers with the following results: PressureSafe demonstrated very high efficacy in noninvasively detecting the presence and absence of PIs below the skin’s surface. PressureSafe accurately detected the presence of a PI in 96% of cases. In addition, PressureSafe correctly determined that no wound was present in 91% of cases. The study was conducted at two medical centers owned by Clalit, the world’s second largest health maintenance organization (HMO) and the largest in Israel, namely Beit Rivka Hospital and Rabin Medical Center both in Petah Tikva, where 370 PressureSafe scans were performed on 25 patients who had Stage 1 PIs or deep tissue injuries. No device related safety issues were reported in the total of 44 patients evaluated for safety.
On September 26, 2023, we announced that we signed a Clinical Trial Agreement with the Methodist Healthcare System of San Antonio to conduct a useability study titled “Safety and Efficacy of the PressureSafe Device for Early Detection of Pressure Injury in People with Various Skin Tones, Including Dark Skin Tones.” Methodist Healthcare is recognized as the most respected healthcare provider in its region. With a network of 85 hospitals, 9 of which are acute care facilities, Methodist Healthcare employs more than 11,000 people, including 2,700 physicians. Based on our intended protocol, we plan to have 50% of the subjects for the upcoming study to have a dark skin tone, thus producing comparative data on PressureSafe’s accuracy as a decision support device in detecting early-stage PIs in people of darker and lighter skin tones. Early-stage PIs can be more difficult to see on dark skin tones with the current standard of care for the detection of PIs, which is visual skin inspection.
| 9 |
On February 20, 2024, we reported 92% efficacy for PressureSafe. Data from the study conducted at two medical centers owned by Clalit, the world’s second largest health maintenance organization (HMO) and the largest in Israel, Beit Rivka Hospital and Rabin Medical Center, presented at the National Pressure Injury Advisory Panel (NPIAP) 2024 Annual Conference on February 16 and 17, 2024 in San Antonio, Texas. The 14-day efficacy portion of the single arm, bi-center study evaluated 38 patients at high risk of pressure injury development. A total of 924 scans were conducted on 154 body locations. Nurses conducting the scans were blinded to PressureSafe’s results, which were encrypted. PressureSafe detected Stage 1 pressure injuries with 92% sensitivity and 88% specificity. Additional portions of the study evaluated safety, as well as device calibration and validation. Total data from 66 patients was obtained for safety analysis and no safety signals were identified in 1,493 scans. Based on these data, the study concluded that PressureSafe is a safe, efficient, and valuable method for early detection of pressure injuries.
On April 9, 2024, the PressureSafe decision support device received an FDA listing certification. PressureSafe is classified as a Class I device and is exempt from 510(k) premarket submission. We are currently working on completing larger scale production of the commercial version of the PressureSafe device, planned to be launched during September 2024, following the listing under the FDA.
DiaSafe
We are now in the development stages of the Software/Hardware, algorithms and optics to allow early detection of incipient DFU in the lower limbs, the DiaSafe. DiaSafe is an adjustment to the PressureSafe proven technology allows us to reduce the development period and approach the relevant markets faster. We plan to initiate a clinical study in Clalit, Israel’s largest HMO, to train the developed algorithm and test patients. Following the Israeli trial, we aim to conduct an American based trial, potentially during the first half of 2025.
DFU Background
Diabetic foot ulcer is an increasing problem among diabetic patients. Diabetic foot ulcers are one of several serious complications of diabetes progression. Major contributing causes to diabetic foot ulcers are peripheral neuropathy, peripheral arterial disease, and immunosuppression. Up to 15% of patients with diabetes have diabetic foot ulcers, and these ulcers lead to more than 80,000 amputations per year in the United States. The lifetime risk of diabetic foot ulcers for patients with diabetes may reach up to 68 per 1,000 persons as reported by some studies. As a diabetic foot ulcer progresses, the patient’s risk for amputation increases; in nearly 84% of patients who have a lower limb amputation secondary to diabetes, the amputation is preceded by a diabetic foot ulcer. Peripheral neuropathy secondary to diabetes is an etiologic factor of diabetic foot ulcers and is estimated to affect 5.5 million people in the United States.
These collective findings indicate that diabetic foot ulcers lead to serious disability, serious reduction in patient quality of life, and high financial costs for society. With increased vigilance on risk assessment, diagnosis, and management of diabetic foot ulcers, clinicians can improve patient outcomes and reduce healthcare costs.
There are a few established methods for diagnosing DFU. These methods assess side effects of diabetic related symptoms as Peripheral Artery Disease diabetic neuropathy (mono-filament test tuning fork test), skin temperature, BP, heart rate, skin dryness etc. The suggested DiaSafe device measures actual dermal and subdermal changes of injured skin tissue structure caused directly the development of diabetic foot ulcers. The optical platform developed by IR-MED allows direct assessment and measurement of changes in skin structure (including blood flow changes).9
| ● | Market Prevalence: The percentage of Americans aged 65 and older diagnosed with diabetes remains high, at 29.2%, or 16.5 million seniors (diagnosed and undiagnosed). |
9 Tuttolomondo A, Maida C, Pinto A. Diabetic foot syndrome: Immune-inflammatory features as possible cardiovascular markers in diabetes. World J Orthop. 2015 Jan 18;6(1):62-76. doi: 10.5312/wjo.v6.i1.62. PMID: 25621212; PMCID: PMC4303791.
10 https://journals.lww.com/jaapa/fulltext/2015/05000/pathogenesis_and_management_of_diabetic_foot.6.aspx
| 10 |
| ● | Diabetic foot ulcers are wounds on the feet that develop in patients with type 1 or type 2 diabetes. About one-third of people with diabetes develop a foot ulcer during their lifetime. Diabetic foot ulcers affect about 18.6 million people worldwide and 1.6 million in the U.S. annually. Treatment of infection in diabetic ulcer is difficult and expensive. Patients usually need to take long-term medications or become hospitalized for an extended period of time DFU treatment is expensive. On average, the treatment cost for wounds with Wagner grade I in five industrialized countries was $3,096 in 2010. However, if the wound becomes complicated and amputated, the cost will rise to almost $107,900.10 Average in-hospital costs were $10,827 (range: $702–$82,880) per DFU episode. Primary healed DFUs costs on average $4,830, single minor amputations on average $13,580, multiple minor amputations on average $31,835 and major amputations on average $73,813 per episode.11 |
All the diabetic patients should undergo comprehensive foot exam once a year. The goal of this examination is to determine the risk factors that may result in foot ulcer and consequently amputation of the affected organ. The physical examination contains observation, palpation of the pulses in the lower extremities, including the posterior tibial and dorsalis pedis pulses. The physical examination also includes neurological tests. At least two neurologic tests are performed and one of the tests should measure the protective sensation in which a 10 g monofilament is used. Vibration sensation using a 128 Hz diapason.
DiaSafe
The DiaSafe device as the PressureSafe device is being designed to capture, analyze and identify tissue status to make early DFU detection and classification using Spectrographic Analysis, while AI based algorithm is implemented to improve provided diagnostic accuracy. The DiaSafe device illuminates the skin with a miniature set of LEDs less than a second in order to acquire the tissue fingerprint. The emitted light photons from the device will be absorbed, scattered and reflected back. The device will then detect the absorption and reflectance, and by using algorithms, it will process the signals to identify and classify the scanned area or DFU.
As all person’s skin properties are unique, the diagnosing physician needs to use a device as the DiaSafe, which automatically calibrate the device to the specific patient’s skin, a process that takes merely a few seconds and allows personalized diagnosis, improving physician diagnostic process effectiveness, as the DiaSafe device is designed to measure regardless of skin color. Our technology is being developed to enable the assessment of different subepidermal layers by scanning through these skin layers, thus improving the identification of the damage, assessing the subepidermal damaged tissue volume and assisting with additional information to allow better treatment efficacy. Measuring the differences of subepidermal biomarker is being developed to detect early formation of DFUs and to “raise a flag” to allow the caregivers intervene and prevent their appearance. The biomarkers that our algorithm detects start from the early inflammatory process, as soon as local underlying tissue function is disturbed, and cells begin to be damaged.
DiaSafe is a hand-held scanner designed to provide additional information as a decision support system (DSS), to support the care giver effectively with the main diagnostic ability to identify DFUs and to differentiate between DFUs under different skin conditions (before they become visible. The DiaSafe is composed of: (a) a handheld optic probe device, which utilizes harmless infra-red light that is placed on the skin and has a disposable tip which is changed between patients. The optic probe with its disposable cover is placed on suspected areas for performing measurements; (b) a disposable probe tip component, changed between patients to avoid cross-contamination; (c) a software component containing machine learning algorithm for analyzing the collected data; and (d) software for connectivity and downloading the collected data and measurements results to the EMR/EHR systems used by the medical center or homecare company.
DiaeSafe is a non-invasive real-time optical monitoring device to support early intervention in DFU treatment prior to skin breakage. The device performs a reflectance spectroscopy scan to generate information for the decision maker, while collecting data on epidermal and subepidermal physiological changes together with other bio-signals typical of early formation of PIs in the main three skin layers, thus detecting the appearance of life risking pressure DiaSafe is designed to detect changes deep in the skin, regardless of skin tone, by measuring bio markers. As soon as local subcutaneous tissue function is disturbed and cells begin to disintegrate by pressure exerted upon the body area, our scanner is designed to be able to detect this very early inflammatory process and tissue structure changes. The technology will allow patient monitoring and immediate reading in a non-invasive way. It has the potential to help to reduce the number of DFUs dramatically through accurate early classification, making it attractive for public and private healthcare systems worldwide.
11 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3634178/ Iraj B, Khorvash F, Ebneshahidi A, Askari G. Prevention of diabetic foot ulcer. Int J Prev Med. 2013;4(3):373-376, https://jamanetwork.com/journals/jama/fullarticle/2812203.
12 https://www.sciencedirect.com/science/article/abs/pii/S0168822717302413.
| 11 |
Recent Developments
On June 4, 2024, we entered into a Securities Purchase Agreement (the “Purchase Agreement”) with certain investors, pursuant to which we agreed to issue and sell, in a private placement offering (the “2024 Private Placement”), 715,000 shares of our Common Stock, at a per share price of $1.00 and warrants to purchase up to an additional 1,144,000 shares of Common Stock at a per share exercise price of $1.00. The 2024 Private Placement closed on June 7, 2024, and we received aggregate gross proceeds of $715,000.
The warrants are exercisable beginning on the six (6) month anniversary of their issuance, have a term of five years from the initial exercise date and entitle the holders to purchase up to 1,144,000 shares of Common Stock. The warrants have an exercise price of $1.00 per share and contain a one-time dilution protection in the event we sell securities at a price less than the then exercise price in effect in a public offering in conjunction with a listing on a national securities exchange.
The securities issued in the 2024 Private Placement were exempt from the registration requirements of the Securities Act of 1933, as amended (the “Securities Act”) pursuant to Section 4(a)(2) of the Securities Act and/or Rule 506(b) of Regulation D promulgated thereunder and pursuant to Regulation S of the Securities Act to non-U.S. investors, because, among other things, the transaction did not involve a public offering, the investors are accredited investors, the investors are taking the securities for investment and not resale and we took appropriate measures to restrict the transfer of the securities. The securities have not been registered under the Securities Act and may not be sold in the United States absent registration or an exemption from registration.
The Purchase Agreement contains representations and warranties that the parties made to, and solely for the benefit of, the others in the context of all of the terms and conditions of that agreement and in the context of the specific relationship between the parties. The investors in the 2024 Private Placement received piggyback registration rights for their shares of Common Stock and shares underlying the associated warrants.
On June 6, 2024, following the approval by its Board and by the Company’s stockholders at the Company’s Annual Meeting held on November 7, 2023, the Company filed a certificate of amendment to the Company’s Articles of Incorporation to increase the number of authorized common shares of the Company from two hundred and fifty million (250,000,000) shares, par value $0.001 per share, to six hundred million (600,000,000) shares, par value $0.001 per share.
Implications of Being a Smaller Reporting Company
We are a “smaller reporting company” as defined in Rule 12b-2 of the Exchange Act and have elected to take advantage of certain scaled disclosure available to smaller reporting companies. As a smaller reporting company, we are eligible and have taken advantage of certain exemptions from various reporting requirements that are not available to public reporting companies that do not qualify for this classification, including, but not limited to:
● An opportunity for reduced disclosure obligations regarding executive compensation in our periodic and annual reports, including without limitation exemption from the requirement to provide a compensation discussion and analysis describing compensation practices and procedures,
● An opportunity for reduced financial statement disclosure in registration statements and in annual reports on Form 10-K, which only requires two years of audited financial statements rather than the three years of audited financial statements that are required for other public companies,
| 12 |
● An opportunity for reduced audit and other compliance expenses as we are not subject to the requirement to obtain an auditor’s report on internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act of 2002, and
● An opportunity to utilize the non-accelerated filer time-line requirements beginning with our annual report for the year ending December 31, 2023, and quarterly filings thereafter.
For as long as we continue to be a smaller reporting company, we expect that we will take advantage of both the reduced internal control audit requirements and the disclosure obligations available to us as a result of this classification.
Risks Associated with Our Business
Our business is subject to numerous risks, as more fully described in the section titled “Risk Factors” to this prospectus. You should read these risks before you invest in our common stock.
Corporate Information
IR-Med, Inc. was incorporated in the state of Nevada on April 20, 2007, under the name “Monster Motors, Inc.” On June 24, 2009, the corporate name was changed to Eco2 Forests, Inc. During September 2012, Eco2 Forests, Inc., accepted a court ordered receiver who authorized a reduction of the authorized shares from 900,000,000 to 500,000,000 and in November 2012 effectuated a 16,000 to 1 stock split. In February 2013, the Company underwent a change of control. On March 25, 2013, Eco2 Forests, Inc., effectuated a 4-to-1 reverse stock split in addition to changing the corporate name to International Display Advertising, Inc. IR-Med, Inc. began operating the business of IR. Med Ltd. An Israeli company, through a reverse acquisition on December 24, 2020 (the “Acquisition”). IR. Med Ltd. (an Israeli company which was founded in 2013) continues as an operating subsidiary of IR-Med, Inc.; IR-Med, Inc. is the sole stockholder of IR. Med Ltd. IR-Med, Inc.’s corporate headquarters and IR. Med Ltd.’s research facilities are located at ZHR Industrial Zone, Rosh Pina, Israel.
| 13 |
THE OFFERING
| Issuer | IR-Med, Inc. | |
| Securities offered by us | $ of Units, each Unit consisting of one share of our common stock and one warrant to purchase one share of our common stock. Each warrant will have an exercise price of $ per share (100% of the public offering price of the common stock), is exercisable immediately and will expire [_] years from the date of issuance. The Units will not be certificated or issued in stand-alone form. The shares of our common stock and the warrants comprising the Units are immediately separable upon issuance and will be issued separately in this offering. | |
| Offering price per Unit | $[_] per Unit | |
| Common stock outstanding prior to this offering* | [_] shares of common stock | |
| Number of shares of common stock offered by us | [_] shares | |
| Number of warrants offered by us | [_] warrants | |
| Common stock to be outstanding immediately after this offering** | [_] shares of common stock (assuming none of the warrants issued in this offering are exercised). | |
| Use of proceeds | We estimate that the net proceeds from this offering will be approximately $ million based on an assumed public offering price of $ per Unit, after deducting estimated underwriting discounts and commissions and estimated offering expenses payable by us.
We currently intend to use the net proceeds from this offering for the development efforts of our products, while mainly focusing on the DiaSafe device, production of commercial units, marketing, and working capital.
See “Use of Proceeds” for additional information. | |
| Representative’s warrants | The registration statement of which this prospectus is a part also registers for sale warrants (the “Representative’s Warrants”) to purchase [_] shares of our common stock ([_] shares of common stock if the over-allotment option is exercised in full) to Maxim Group LLC (the “Representative”), as the representative of the several underwriters, as a portion of the underwriting compensation payable to the underwriters in connection with this offering. The Representative’s Warrants will be exercisable commencing six months following the effective date of the registration statement of which this prospectus is a part and expiring on the fifth anniversary of the commencement of sales of this offering at an exercise price of $[_] (110% of the public offering price of the Units). Please see “Underwriting — Representative’s Warrants” for a description of these warrants. |
| 14 |
| Over-allotment option | The Underwriting Agreement provides that we will grant to the underwriters an option, exercisable within [_] days after the closing of this offering, to acquire up to an additional [_]% of the total number of shares of common stock and/or warrants to be offered by us pursuant to this offering, solely for the purpose of covering over-allotments. | |
| Lock-Up | Our directors, executive officers, and certain stockholders have agreed with the Representative not to offer for sale, issue, sell, contract to sell, pledge or otherwise dispose of any of our common stock or securities convertible into common stock for a period of 6 months commencing on the date of this prospectus. | |
| Risk Factors | You should read the section titled “Risk Factors” for a discussion of factors to consider carefully, together with all the other information included in this prospectus, before deciding to invest in our common stock. | |
| Proposed Nasdaq Capital Market listing | Our common stock is currently quoted on the OTCQB Market. We intend to apply to list our common stock and warrants on Nasdaq under the symbols “IRME.” and “IRMEW”, respectively. If our application is not approved or we otherwise determine that we will not be able to secure the listing of our common stock on Nasdaq, we will not complete this offering. |
*The number of shares of our common stock to be outstanding immediately after this offering is based on 70,699,144 shares of common stock outstanding as of June 24, 2024 and excludes:
| ● | 13,924,175 shares of our common stock issuable upon the exercise of stock options as of June 17, 2024, at a weighted-average exercise price of $0.42 per share; | |
| ● | 12,474,259 shares of our common stock issuable upon the exercise of warrants as of June 17, 2024; and | |
| ● | 3,575,825 shares of our common stock that remain available for issuance as of June 17, 2024 under our 2020 Incentive Stock Plan. |
Unless otherwise indicated, all information contained in this prospectus assumes or gives effect to:
| ● | no exercise of the warrants included in the Units; | |
| ● | no exercise of the underwriters’ option to purchase additional shares and/or warrants; and | |
| ● | no exercise by the Underwriter of its option to purchase warrants to purchase six (6) percent of the common stock sold in this offering as set forth on the cover page of this prospectus, that will be issued to the Underwriter in connection with this offering (the “Underwriter Warrants”). |
| 15 |
FINANCIAL DATA
The following tables set forth our summary consolidated statements of operations and consolidated balance sheet data. The summary consolidated statements of operations data for the years ended December 31, 2023 and 2022 and the consolidated balance sheet data as of December 31, 2023 are derived from our audited consolidated financial statements appearing elsewhere in this prospectus. The summary of the interim unaudited consolidated balance sheet data for the three months ended March 31, 2024, are derived from our interim unaudited consolidated financial statements appearing elsewhere in this prospectus. Our historical results are not necessarily indicative of the results that may be expected for any period in the future and our interim results are not necessarily indicative of our expected results for the year ending December 31, 2024. You should read the following summary consolidated financial data together with the section titled “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and our consolidated financial statements and the related notes included elsewhere in this prospectus.
Statement of Operations Data
U.S. dollars (in thousands)
| For the year | For the year | |||||||
| ended | ended | |||||||
| December 31, | December 31, | |||||||
| 2023 | 2022 | |||||||
| US Dollars (In thousands) | ||||||||
| Research and development expenses | 2,061 | 1,885 | ||||||
| Marketing expenses | 822 | 759 | ||||||
| General and administrative expenses | 2028 | 2,118 | ||||||
| Total operating loss | 4,911 | 4,762 | ||||||
| Financial income, net | (2 | ) | (28 | ) | ||||
| Loss for the year | 4,909 | 4,734 | ||||||
| Loss per share | ||||||||
| Basic and dilutive loss per common stock (in U.S. dollars) | (0.07 | ) | (0.07 | ) | ||||
| 16 |
| For the year | For the year | |||||||
| ended | ended | |||||||
| December 31, | December 31, | |||||||
| 2023 | 2022 | |||||||
| US Dollars (In thousands) | ||||||||
| Cash flows from operating activities | ||||||||
| Loss for the year | (4,909 | ) | (4,734 | ) | ||||
| Adjustments to reconcile loss for the year to net cash used in operating activities: | ||||||||
| Stock based compensation | 1,682 | 1,330 | ||||||
| Depreciation | 17 | 10 | ||||||
| Accrued financial income | (3 | ) | (28 | ) | ||||
| Decrease (increase) in accounts receivable | (25 | ) | 12 | |||||
| Increase in trade and other payables | 6 | 35 | ||||||
| Net cash used in operating activities | (3,232 | ) | (3,375 | ) | ||||
| Cash flows from investing activities | ||||||||
| Purchase of property and equipment | (2 | ) | (50 | ) | ||||
| Increase in long term deposit | - | (9 | ) | |||||
| Net cash used in investing activities | (2 | ) | (59 | ) | ||||
| Cash flows from financing activities | ||||||||
| Proceeds from private placement of common stock and warrants. | 1,000 | 3,625 | ||||||
| Net cash provided by financing activities | 1,000 | 3,625 | ||||||
| Effect of exchange rate changes on cash | (1 | ) | (4 | ) | ||||
| Net (decrease) increase in cash and cash equivalents | (2,235 | ) | 187 | |||||
| Cash and cash equivalents as at the beginning of the year | 3,002 | 2,815 | ||||||
| Cash and cash equivalents as at the end of the year | 767 | 3,002 | ||||||
| Non-cash financing Activities: | ||||||||
| Increase in right of use assets against lease liability | - | 155 | ||||||
| Increase in right of use assets against long term restricted deposits | - | 19 | ||||||
Balance
Sheet Data
(in thousands, except share and per share data)
| 17 |
| 2023 | 2022 | |||||||
| US Dollars (In thousands) | ||||||||
| Assets | ||||||||
| Current assets | ||||||||
| Cash and cash equivalents | 767 | 3,002 | ||||||
| Accounts receivable | 81 | 55 | ||||||
| Total current assets | 848 | 3,057 | ||||||
| Non-current assets | ||||||||
| Long term restricted deposits | 11 | 11 | ||||||
| Right of use assets | 84 | 155 | ||||||
| Property and equipment, net | 56 | 71 | ||||||
| Total non-current assets | 151 | 237 | ||||||
| Total assets | 999 | 3,294 | ||||||
| Liabilities and stockholders’ equity | ||||||||
| Current liabilities | ||||||||
| Trade and other payables | 473 | 500 | ||||||
| Stockholders’ loans | - | 162 | ||||||
| Total current liabilities | 473 | 662 | ||||||
| Non-current liabilities | ||||||||
| Stockholders’ loans | 161 | - | ||||||
| Long term lease liability | - | 40 | ||||||
| Total non-current liabilities | 161 | 40 | ||||||
| Total liabilities | 634 | 702 | ||||||
| Contingent liabilities and commitments | ||||||||
| Stockholders’ equity | ||||||||
| Common stock, par value $0.001 per share, 250,000,000 shares authorized: 69,931,056 and 68,808,970 issued and outstanding as of December 31, 2023, and 2022, respectively | 69 | 68 | ||||||
| Additional paid-in capital | 15,135 | 12,454 | ||||||
| Accumulated deficit | (14,839 | ) | (9,930 | ) | ||||
| Total stockholders’ equity | 365 | 2,592 | ||||||
| Total liabilities and stockholders’ equity | 999 | 3,294 | ||||||
| 18 |
Interim Unaudited Condensed Consolidated Balance Sheets
| March 31 2024 | December 31 2023 | |||||||
| USD thousands | USD thousands | |||||||
| Assets | ||||||||
| Current assets | ||||||||
| Cash and cash equivalents | 408 | 767 | ||||||
| Accounts receivable | 55 | 81 | ||||||
| Total current assets | 463 | 848 | ||||||
| Non- current assets | ||||||||
| Long term restricted deposit | 11 | 11 | ||||||
| Right of use assets | 58 | 84 | ||||||
| Property and equipment, net | 47 | 56 | ||||||
| Total non-current assets | 116 | 151 | ||||||
| Total assets | 579 | 999 | ||||||
| Liabilities and stockholders’ equity (deficiency) | ||||||||
| Current liabilities | ||||||||
| Trade and other payables | 505 | 473 | ||||||
| Non-current liabilities | ||||||||
| Stockholders’ loans | 160 | 161 | ||||||
| Total liabilities | 665 | 634 | ||||||
| Stockholders’ equity (deficiency) | ||||||||
| Common Stock, par value $0.001 per share, 250,000,000, shares authorized. As of March 31, 2024, and December 31, 2023, 69,931,056 shares were issued. | 69 | 69 | ||||||
| Additional paid-in capital | 15,341 | 15,135 | ||||||
| Accumulated deficit | (15,496 | ) | (14,839 | ) | ||||
| Total Stockholders’ equity (deficiency) | (86 | ) | 365 | |||||
| Total liabilities and stockholders’ equity (deficiency) | 579 | 999 | ||||||
| 19 |
Interim Unaudited Condensed Consolidated Statements of Operations
For the three-months period ended March 31 | ||||||||
| 2024 | 2023 | |||||||
| U.S dollars (in thousands) | ||||||||
| Research and development expenses: | ||||||||
| Expenses incurred | 375 | 605 | ||||||
| Less- government participation | (180 | ) | - | |||||
| Research and development expenses, net | 195 | 605 | ||||||
| Marketing expenses | 168 | 172 | ||||||
| General and administrative expenses | 295 | 575 | ||||||
| Total operating loss | 658 | 1,352 | ||||||
| Financial income, net | (1 | ) | (2 | ) | ||||
| Loss for the period | 657 | 1,350 | ||||||
| Basic and dilutive loss per common stock (in dollars) | (0.01 | ) | (0.02 | ) | ||||
Weighted-average shares in the loss per share computation for the three months ended March 31, 2024 and, 2023 were 69,931,056 and 68,829,424 respectively.
The accompanying notes are an integral part of these interim unaudited condensed consolidated financial statements.
Interim Unaudited Condensed Consolidated Statements of Cash Flows
| For the three-month period ended | ||||||||
| March 31 | March 31 | |||||||
| 2024 | 2023 | |||||||
| U.S dollars (in thousands) | ||||||||
| Cash flows from operating activities | ||||||||
| Loss for the period | (657 | ) | (1,350 | ) | ||||
| Adjustments to reconcile loss for the period to net cash used in operating activities: | ||||||||
| Stock based compensation | 206 | 478 | ||||||
| Depreciation | 9 | 4 | ||||||
| Accrued financial expenses (income) | 3 | (6 | ) | |||||
| Decrease (increase) in accounts receivable | 27 | (1 | ) | |||||
| Increase (decrease) in trade and other payables | 54 | (25 | ) | |||||
| Net cash used in operating activities | (358 | ) | (900 | ) | ||||
| Effect of exchange rate changes on cash and cash equivalents | (1 | ) | 1 | |||||
| Net decrease in cash and cash equivalents | (359 | ) | (899 | ) | ||||
| Cash and cash equivalents as at the beginning of the period | 767 | 3,002 | ||||||
| Cash and cash equivalents as at the end of the period | 408 | 2,103 | ||||||
The accompanying notes are an integral part of these interim unaudited condensed consolidated financial statements.
| 20 |
RISK FACTORS
Investing in our securities involves a high degree of risk. You should carefully consider the risks described below, together with the other information contained in this prospectus, including our consolidated financial statements and the related notes, before making any decision to invest in securities. This prospectus contains forward-looking statements. If any of the events discussed in the risk factors below occurs, our business, prospects, results of operations, financial condition and cash flows could be materially harmed. If that were to happen, the trading price of our common stock could decline, and you could lose all or part of your investment. The risks and uncertainties described below are not the only ones we face. Additional risks not currently known to us, or other factors not perceived by us to present significant risks to our business at this time also may impair our business operations.
Summary of Risk Factors
Below is a summary of the principal factors that make an investment in our securities speculative or risky. This summary does not address all of the risks that we face. Additional discussion of the risks summarized in this risk factor summary, and other risks that we face, can be found below under the heading “Risk Factors” and should be carefully considered, together with other information in this prospectus and our other filings with the SEC, before making an investment decision regarding our securities.
● We are a development stage medical device company and have a history of significant operating losses; we expect to continue to incur operating losses, and we may never achieve or maintain profitability.
● We will need substantial additional funding to continue our operations, which could result in significant dilution or restrictions on our business activities. We may not be able to raise capital when needed, if at all, which would force us to delay, reduce or eliminate our product development programs or commercialization efforts and could cause our business to fail.
● Our independent registered public accounting firm has included an explanatory paragraph relating to our ability to continue as a going concern in its report on our audited financial statements included in this report. Our audited financial statements on December 31, 2023, were prepared assuming that we will continue as a going concern.
● Medical device development involves a lengthy and expensive process with an uncertain outcome. We may incur additional costs or experience delays in completing, or ultimately be unable to complete, the development and commercialization of any product.
● We currently have no products that are approved for commercial sale. If we are unable to successfully develop, receive commercial sale approval from the regulatory authorities as applicable and commercialize initially our PressureSafe device under development, or if we experience significant delays in doing so, our business will be adversely affected.
● The size and future growth in the market for our planned devices under development has not been established with precision and may be smaller than we estimate, possibly materially. If our estimates and projections overestimate the size of this market, our sales growth may be adversely affected.
● Any product candidates we may advance into clinical trials (assuming the FDA so requires) may be subject to extensive regulation, which can be costly and time consuming, cause unanticipated delays or prevent the receipt of the required approvals to commercialize our product candidates, all of which can adversely affect our business
● We may be subject to numerous and varying privacy and security laws, and our failure to comply could result in penalties and reputational damage.
● If we are unable to establish sales and marketing capabilities or fail to enter into agreements with third parties to market and sell any products we may successfully develop, we may not be able to effectively market and sell any such products and generate product revenue.
● Failure to manage our growth effectively could increase our expenses, decrease our revenue and prevent us from implementing our business strategy.
● Failure to secure or retain coverage or adequate reimbursement for our planned products in development by third-party payors could adversely affect our business, financial condition and operating results.
| 21 |
● If we are unable to protect our intellectual property rights or if our intellectual property rights are inadequate to protect our technology, our competitors could develop and commercialize technology similar to ours, and our competitive position could be harmed.
● Our technology development is headquartered in Israel and, therefore, our results may be adversely affected by economic restrictions imposed on, and political and military instability in Israel, including the Israel-Hamas war.
● There is not now, and there may never be, an active, liquid and orderly trading market for our common stock, which may make it difficult for you to sell your shares of our common stock.
● Directors, executive officers, principal stockholders and affiliated entities own a significant percentage of our capital stock, and they may make decisions that our stockholders do not consider to be in their best interests.
● Sale of our common stock by our stockholders could encourage short sales by third parties, which could contribute to the further decline of our stock price.
● There is a limited existing market for our common stock.
● Our share price is expected to be volatile and may be influenced by numerous factors, some of which are beyond our control.
● If securities or industry analysts do not publish, or cease publishing, research or publish inaccurate or unfavorable research about our business or our market, or if they change their recommendations regarding our stock adversely, our stock price and any trading volume could decline.
● We may have become exposed to material liabilities that were not discovered before, and have not been discovered since, due to the closing of the Acquisition.
● We are exposed to additional risks as a result of “going public” by means of a reverse acquisition transaction.
● If we fail to maintain proper and effective internal controls, our ability to produce accurate and timely financial statements could be impaired, which could harm our operating results, our ability to operate our business and investors’ views of us.
● Shares of our common stock that have not been registered under federal securities laws are subject to resale restrictions imposed by Rule 144, including those set forth in Rule 144(i) which apply to a former “shell company.”
● If we issue additional shares of our capital stock in the future, our existing stockholders will be diluted.
● Sales of a substantial number of shares of our common stock in the public market, or the perception that such sales could occur, could cause our stock price to fall.
● Anti-takeover provisions in our organizational documents could delay or prevent a change of control.
● Article XI of our Second Amended and Restated Articles of Incorporation designates the Eighth Judicial District Court of Clark County, Nevada as the sole and exclusive forum for certain types of actions and proceedings that may be initiated by our shareholders and therefore may limit our shareholders’ ability to choose a forum for disputes with us or our directors, officers, employees, or agents.
● The elimination of personal liability of our directors and officers under Nevada law and the existence of indemnification rights held by our directors, officers and employees may result in substantial expenses.
● If, after being listing on Nasdaq, we are delisted and our shares become subject to the penny stock rules, it would become more difficult to trade our shares.
● If our securities are listed on Nasdaq, our failure to meet the continued listing requirements of Nasdaq could result in a de-listing of our securities.
● We do not intend to pay cash dividends on our capital stock in the foreseeable future.
● Future sales of shares by existing stockholders could cause our share price to decline.
● Investors in this offering will experience immediate and substantial dilution of dollars per share.
● If securities or industry analysts do not publish research or publish inaccurate or unfavorable research about our business, our stock price and trading volume could decline.
● Provisions in our corporate charter documents and under Nevada law could discourage another company from acquiring us and may prevent attempts by our stockholders to replace or remove our current management.
| 22 |
Risk Factors
Risks Related to Our Business, Financial Position, Capital Requirements, Managing our Growth and Other Legal Compliance Matters
We are a development stage medical device company and have a history of significant operating losses; we expect to continue to incur operating losses, and we may never achieve or maintain profitability.
As a development stage company, we do not currently have revenues to generate cash flows to cover operating expenses. Since our inception, we have incurred operating losses in each year due to costs incurred in connection with research and development activities, marketing and general and administrative expenses associated with our operations. For the years ended December 31, 2023, and 2022, we incurred net losses of approximately $4,909,000 and $4,734,000, respectively. As of December 31, 2023, and 2022, we had an accumulated deficit of $14,839,000 and $9,930,000, respectively.
We expect to incur losses for the foreseeable future as we continue the development of, and seek regulatory clearance and approvals for, our devices-in-development initially (for pre-emptive diagnosis of PIs on the skin surface), DiaSafe and thereafter, for the future Nobiotics device (for detecting the ear infections in children). If we fail to generate revenue and eventually become profitable, or if we are unable to fund our continuing losses, our shareholders could lose all or a substantial part of their investment.
We will need substantial additional funding to complete subsequent phases of our medical devices and to operate our business and such funding may not be available or, if it is available, such financing is likely to substantially dilute our existing shareholders.
The discovery, development and commercialization of new medical devices (such as our PressureSafe and Diasafe devices) entails significant costs. As we are in the stage of the engineering, electronics, algorithm and mechanical aspects of our devices and prototypes, we still must develop, modify, refine and finalize them. To enable us to accomplish these and other related items and continue to operate our business, we will need to raise substantial additional capital or enter into strategic partnerships to enable us to:
| ● | fund clinical studies and seek regulatory approvals/clearance prior to performing clinical trials (if needed); | |
| ● | build or access manufacturing and commercialization capabilities; | |
| ● | develop, test and receive regulatory commercial sale approval to market our products; | |
| ● | acquire or license additional internal systems and other infrastructure; and | |
| ● | hire and support additional management, engineering and scientific personnel. |
We will need substantial additional funding to continue our operations, which could result in significant dilution or restrictions on our business activities. We may not be able to raise capital when needed, if at all, which would force us to delay, reduce or eliminate our product development programs or commercialization efforts and could cause our business to fail.
Our operations have consumed substantial amounts of cash since inception. We expect to need substantial additional funding to pursue the clinical development of our drug candidates and launch and commercialize any drug candidates for which we receive regulatory approval.
In 2021 and 2022, we raised aggregate gross proceeds of $5,830,000 and $3,625,000, respectively, from sales of our equity and equity linked securities. On June 12, 2023, we raised aggregate gross proceeds of $1,000,000 from sales of our shares of common stock and warrants to purchase shares of common stock. On June 4, 2024, we raised aggregate gross proceeds of $715,000 from sales of our shares of common stock and warrants to purchase shares of common stock.
| 23 |
Nonetheless, we will require additional capital for the further development and commercialization of our three product candidates (which are in various stages of design and development) and may need to raise additional funds sooner if we choose to and are able to expand more rapidly than we currently anticipate. Further, we expect our expenses to increase in connection with our ongoing activities. In addition, if we obtain regulatory approval for any of our product candidates, we expect to incur significant commercialization expenses related to regulatory requirements, product manufacturing, marketing, sales and distribution.
Furthermore, we expect to incur additional costs associated with operating as a public company. We may also encounter unforeseen expenses, difficulties, complications, delays and other unknown factors that may increase our capital needs and/or cause us to spend our cash resources faster than we expect. Accordingly, we will need to obtain substantial additional funding in order to continue our operations.
To date, we have financed our operations through a mix of equity investments from private investors, the incurrence of debt, grant funding and technology licensing revenues, and we expect to continue to utilize such means of financing for the foreseeable future. Additional funding from those or other sources may not be available when or in the amounts needed, on acceptable terms, or at all.
If we raise capital through the sale of equity, or securities convertible into equity, it will result in dilution to our then existing stockholders, which could be significant depending on the price at which we may be able to sell our securities.
If we raise additional capital through the incurrence of indebtedness, we may become subject to covenants restricting our business activities, and holders of debt instruments may have rights and privileges senior to those of our equity investors. In addition, servicing the interest and principal repayment obligations under debt facilities could divert funds that would otherwise be available to support research and development or commercialization activities.
If we are unable to raise capital when needed on commercially reasonable terms, we could be forced to delay, reduce or eliminate our research and development for our product candidates or any future commercialization efforts or ultimately cease operations. Any of these events could significantly harm our business, financial condition and prospects.
Until we can generate a sufficient amount of product revenue to finance our cash requirements, which we may never achieve, we expect to finance our cash needs primarily through public or private equity offerings, debt financings or through the establishment of possible strategic alliances. We cannot be certain that additional funding will be available on acceptable terms, or at all. If we are not able to secure additional equity funding when needed, we may have to delay, reduce the scope of, or eliminate one or more of our clinical studies, development programs or future commercialization initiatives.
In addition, any additional equity funding that we do obtain will dilute the ownership held by our existing security holders. The amount of this dilution may be substantially increased if the trading price of our common stock is lower at the time of any financing. Regardless, the economic dilution to shareholders will be significant if our stock price does not increase significantly, or if the effective price of any sale is below the price paid by a particular shareholder. Any debt financing that we obtain in the future could involve substantial restrictions on activities and creditors could seek a pledge of some or all of our assets. We have not identified potential sources for such financing that we will require, and we do not have commitments from any third parties to provide any future debt financing. If we fail to obtain funding as needed, we may be forced to cease or scale back operations, and our results, financial condition and stock price would be adversely affected.
| 24 |
Our independent registered public accounting firm has included an explanatory paragraph relating to our ability to continue as a going concern in its report on our audited financial statements included in this report. Our audited financial statements on December 31, 2023, were prepared assuming that we will continue as a going concern.
Primarily as a result of our losses and limited cash balances and cash flows, the report of our independent registered public accounting firm for the fiscal year ended December 31, 2023, contains an explanatory paragraph on our financial statements stating that the Company has not generated sufficient revenues to cover operating expenses and will need additional capital to service its debt obligations. The Company raised proceeds of $1,000,000 during the year ended December 31, 2023, by way of private placement offerings to accredited investors, following this offering we believe we will have sufficient resources to meet our operating and capital needs for the next four months. The Company expects it will require additional capital to fully implement the scope of its proposed business operations, which raises substantial doubt about its ability to continue as a going concern. The Company will have to continue to rely on equity and debt financing, and/or continued support from its officers and directors. There can be no assurance that financing, whether debt or equity, will be available to the Company in the amount required at any particular time or for any particular period or, if available, that it can be obtained on favorable terms.
If we are unable to secure additional capital, we may be required to curtail our clinical and research and development initiatives and take additional measures to reduce costs in order to conserve our cash in amounts sufficient to sustain operations and meet our obligations. These measures could cause significant delays in our clinical and regulatory efforts, which is critical to the realization of our business plan. The accompanying financial statements do not include any adjustments that may be necessary should we be unable to continue as a going concern. It is not possible for us to predict at this time the potential success of our business. The revenue and income potential of our proposed business and operations are currently unknown. If we cannot continue as a viable entity, you may lose some or all of your investment.
Our limited operating history as a development stage company may hinder our ability to successfully meet our objectives.
We were formed in 2013, and since that time our focus has been on our three leading product candidates, which are the PressureSafe device, DiaSafe device. We have limited experience with development stage operations, including manufacturing and have not yet demonstrated an ability to successfully overcome many of the risks and uncertainties frequently encountered by companies in new and rapidly evolving fields, particularly in the medical device support systems arena. In addition, the early-stage nature of our development operations can only provide limited operating results upon which investors can evaluate our business and prospects.
Our limited operating history may adversely affect our ability to implement our business strategy and achieve our business goals, which include, among others, the following activities:
| ● | developing our product candidates using unproven technologies; | |
| ● | undertaking preclinical development and clinical trials as well as formulating and manufacturing products; | |
| ● | obtaining the human, financial and other resources necessary to develop, test, manufacture, commercialize and market our product candidates; | |
| ● | engaging collaborators to assist in developing, testing, manufacturing and marketing our product candidates; | |
| ● | continuing to build and maintain an intellectual property portfolio covering our technology and product candidates; | |
| ● | achieving acceptance and use by the medical community of our Anticalin platform and drug candidates after they receive regulatory approvals; | |
| ● | maintaining, growing and managing our internal teams as and to the extent we increase our operations and develop new segments of our business; | |
| ● | developing and maintaining successful collaboration, strategic and other relationships for the development and commercialization of our product candidates that receive regulatory approvals with existing and new partners; and | |
| ● | managing our cash flows and any growth we may experience in an environment where costs and expenses relating to clinical studies, regulatory approvals and commercialization continue to increase. |
| 25 |
If we are unsuccessful in accomplishing any or all of these objectives, we may not be able to raise capital, expand our business, develop our drug candidates or continue our operations.
We may never achieve profitability.
Because of the numerous risks and uncertainties associated with the development and commercialization of medical device solutions, we are unable to accurately predict the timing or amount of future revenue or expenses or when, or if, we will be able to achieve profitability. We have financed our operations primarily through issuance and sale of equity and equity linked securities. The size of our future net losses will depend, in part, on the rate of growth or contraction of our expenses and the level and rate of growth, if any, of our revenues. We expect to continue to expend substantial financial and other resources on, among other things:
| ● | investments to expand and enhance our platform and technology infrastructure, make improvements to the scalability, availability and security of our platform, and develop new products; | |
| ● | sales and marketing, including expanding our indirect sales organization and marketing programs; | |
| ● | planning and conducting clinical trials to obtain regulatory approval/clearance for the commercialization of our products; | |
| ● | expansion of our operations and infrastructure, both domestically and internationally; and | |
| ● | general administration, including legal, accounting and other expenses related to being a public company. |
If we are unable to successfully commercialize our products or if revenue from any of our products that receives marketing approval is insufficient, we will not achieve profitability. Furthermore, even if we successfully commercialize our products, our planned investments may not result in increased revenue or growth of our business. We may not be able to generate net revenues sufficient to offset our expected cost increases and planned investments in our business and platform. As a result, we may incur significant losses for the foreseeable future, and may not be able to achieve and sustain profitability. If we fail to achieve and sustain profitability, then we may not be able to achieve our business plan, fund our business or continue as a going concern.
Our quarterly results may fluctuate significantly, and period-to-period comparisons of our results may not be meaningful.
Our quarterly results, including the levels of future revenue, if any, our operating expenses and other costs, and our operating margins, may fluctuate significantly in the future, and period-to-period comparisons of our results may not be meaningful. This may be especially true to the extent that we do not successfully establish our business model. Accordingly, the results of any one period should not be relied upon as an indication of our future performance. In addition, our quarterly results may not fully reflect the underlying performance of our business. Factors that may cause fluctuations in our quarterly results include, but are not limited to:
| ● | the timing of regulatory commercial sale approvals for our products in various stages of development; |
| 26 |
| ● | our ability to successfully establish our business model; | |
| ● | our ability to attract and retain distribution networks, customers and to expand our business; | |
| ● | enacted or pending legislation effecting the healthcare industry; | |
| ● | changes in our pricing policies or those of our competitors; | |
| ● | the timing of our recognition of revenue and the mix of our revenues during the period; | |
| ● | the amount and timing of operating expenses and other costs related to the maintenance and expansion of our business, infrastructure and operations; | |
| ● | the amount and timing of operating expenses and other costs related to the development or acquisition of businesses, services, technologies or intellectual property rights; | |
| ● | the timing and costs associated with legal or regulatory actions; | |
| ● | changes in the competitive dynamics of our industry, including consolidation among competitors or customers; | |
| ● | loss of our executive officers or other key employees | |
| ● | industry conditions and trends that are specific to the vertical markets in which we sell or intend to sell our devices; and | |
| ● | general economic and market conditions. |
Fluctuations in quarterly results may negatively impact the value of our common stock, regardless of whether they impact or reflect the overall performance of our business. If our quarterly results fall below the expectations of investors or any securities analysts who follow our shares, or below any guidance we may provide, the price of our ordinary shares could decline substantially.
Currency exchange rate fluctuations affect our results of operations, as reported in our financial statements.
We incur expenses in U.S. Dollars and in NIS, but our functional currency is the U.S. dollar. However, a significant portion of our headcount related expenses, consisting principally of salaries and related personnel expenses as well as R&D consulting services, leases and certain other operating expenses, are denominated in NIS. This foreign currency exposure gives rise to market risk associated with exchange rate movements of the U.S. dollar against the NIS. Furthermore, we anticipate that a material portion of our expenses will continue to be denominated in NIS.
In addition, increased international sales in the future may result in greater foreign currency denominated sales, increasing our foreign currency risk. If we are not able to successfully hedge against the risks associated with currency fluctuations, our financial condition and results of operations could be adversely affected.
| 27 |
Medical device development involves a lengthy and expensive process with an uncertain outcome. We may incur additional costs or experience delays in completing, or ultimately be unable to complete, the development and commercialization of any product.
Before the DiaSafe and the Nobiotics medical devices can be available for commercial sale the United States and in other countries, we must complete all regulatory requirements necessitated by the FDA and foreign health regulatory authorities and demonstrate the performance and safety of our technology. These activities will include performing clinical useability studies. While we currently plan to pursue 510(k) approval which does not require clinical trials, the FDA may require clinical trials in order to approve our product candidates. Clinical Trials are expensive, difficult to design and implement, can take years to complete and are inherently uncertain as to outcome. A failure of one or more clinical trials can occur at any stage of testing. Further, the outcomes of completed clinical trials may not be predictive of the success of later clinical trials, and interim results of a clinical trial do not necessarily predict final results. Clinical data is often susceptible to varying interpretations and analyses, and many companies that have believed their products performed satisfactorily in clinical trials have nonetheless failed to obtain marketing approval. We have limited resources to complete the expensive process of medical device development, and clinical trials, putting us at a disadvantage, particularly compared to some of our larger and established competitors, and we may not have sufficient resources to commercialize our products under development in a timely fashion, if ever.
We may experience numerous unforeseen events during or as a result of clinical trials that we may be required to perform that could delay or prevent our ability to receive marketing approval or commercialize our products, including:
| ● | regulators may not authorize us or our investigators to commence a clinical trial or conduct a clinical trial at a prospective trial site; | |
| ● | the failure to complete clinical trials testing requirements required by the FDA and foreign health regulatory authorities; | |
| ● | we may experience delays in reaching agreement (or fail in reaching agreement) on acceptable clinical trial contracts with third parties or acceptable clinical trial protocols with prospective trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among trial sites; | |
| ● | clinical trials of the technology underlying PressureSafe or DiaSafe devices may produce negative or inconclusive results, including failure to demonstrate statistical significance, and we may decide, or regulators may require us, to conduct additional clinical trials or abandon our development programs; | |
| ● | the number of people with the necessary disorders required for clinical trials may be larger than we anticipate. Enrollment in these clinical trials may be slower than we anticipate. People may drop out of these clinical trials or fail to return for follow-up at a higher rate than we anticipate; | |
| ● | our third-party contractors conducting the clinical trials may fail to comply with regulatory requirements or meet their contractual obligations to us in a timely manner, or at all; | |
| ● | the cost of clinical trials of our products may be greater than we anticipate; | |
| ● | the supply or quality of our products or other materials necessary to conduct clinical trials of our products may be insufficient or inadequate; and | |
| ● | delays from our suppliers and manufacturers could impact clinical trial completion and impact future revenue. |
If we are required to conduct clinical trials or other testing of our proposed devices under development beyond those that we contemplate or if the results of these trials or tests are not favorable or if there are safety concerns, we may:
| ● | not obtain commercial sale approvals at all; | |
| ● | be delayed in obtaining commercial sale approvals for our planned products under development in a jurisdiction; or | |
| ● | be subject to additional testing requirements. |
Our development costs will also increase if we experience delays in testing or commercial sale approval from regulatory authorities. We do not know whether any of our clinical trials will begin as planned, will need to be restructured or will be completed on schedule, or at all. Significant clinical trial delays also could allow our competitors to bring innovative products to market before we do and impair our ability to successfully commercialize our products.
| 28 |
Changes in the configuration of the technology underlying our devices under development may result in additional costs or delay.
As products are developed through towards approval and commercialization, it is common that various aspects of the development program, such as manufacturing methods and configuration, are altered along the way in an effort to optimize processes and results. Any changes we make carry the risk that they will not achieve the intended objectives. Any of these changes could cause our products under development to perform differently and affect the results of planned clinical trials or other future clinical trials conducted with the altered device. Such changes may also require additional testing, regulatory notification or regulatory approval. This could delay completion of clinical trials, increase costs, delay approval of our future products and jeopardize our ability to commence sales and generate revenue.
We currently have no products that are approved for commercial sale. If we are unable to successfully develop, receive commercial sale approval from the regulatory authorities as applicable and commercialize initially our PressureSafe device under development, or if we experience significant delays in doing so, our business will be adversely affected.
We currently have one product that is approved for commercial sale. On April 9, 2024, the PressureSafe decision support device received an FDA listing certification. PressureSafe is classified as a Class I device. We are currently working on completing larger scale production of the commercial version of the PressureSafe device, planned to be launched during September 2024, following the listing under the FDA. Our ability to generate revenue from our developed products, if any, will depend heavily on their successful development, commercial sale approval, and eventual commercialization. The success of any product that we develop will depend on several factors, including:
| ● | receipt of timely approval from foreign health regulatory authorities (if we seek approval in any jurisdiction outside the United States); | |
| ● | successful completion of all necessary bench testing, and clinical trials, if necessary; | |
| ● | our ability to procure and maintain suppliers and manufacturers of the components of the technology underlying PressureSafe and future versions; | |
| ● | launching commercial sales of our devices, if approved for commercial sale; | |
| ● | market acceptance of our devices under development, if approved, by the medical community and third-party payers; | |
| ● | our ability to obtain extensive coverage and reimbursement for use of our devices; | |
| ● | the perceived advantages, cost, safety, convenience and accuracy of alternative diagnostic methods; | |
| ● | obtaining and maintaining patent, trademark and trade secret protection and regulatory exclusivity for our technology and otherwise protecting our rights in our intellectual property portfolio; and | |
| ● | maintaining compliance with regulatory requirements, including current good manufacturing practices. |
Whether FDA commercial sale approval will be granted for additional products is unpredictable and may depend upon several factors, including the substantial discretion of the regulatory authorities. We may need to perform clinical trials, and the FDA (and as we seek to commercialize in selected international geographies, other foreign regulatory authorities) may require that we conduct additional bench testing, and/or clinical trials, provide additional data, take additional manufacturing steps, or require other conditions, before they will let us to market our device. If the FDA or other foreign regulatory authority will require additional clinical trials or data, we would incur increased costs and delays in the access to market, which may require us to expend more resources than we have available.
In cases where we are successful in obtaining commercial sale approval to market one or more of our products, our revenue will be dependent, in part, upon the size of the markets in the territories for which we gain commercial sale approval, the accepted price for the product, the ability to obtain coverage and reimbursement, and whether we own the commercial rights for that territory. If the number of people we target is not as significant as we estimate or the treatment population is narrowed by competition, physician choice or diagnostic guidelines, we may not generate significant revenue from sales of such products, even if they are available on the market.
Commercial sale approval in the United States by the FDA does not guarantee approval by other regulatory authorities in other countries or jurisdictions or ensure approval for the same conditions of use. In addition, clinical trials conducted in one country may not be accepted by regulatory authorities in other countries. Approval processes vary between countries and can involve additional product testing and validation and additional administrative review periods. No product we develop may ever obtain commercial sale approval in the United States or any other jurisdiction, even if we expend substantial time and resources seeking such approval. If we do not achieve one or more of these approvals promptly or at all, we could experience significant delays or an inability to fully commercialize any product and achieve profitability.
| 29 |
Both before and after a product is commercially released, we will have ongoing responsibilities under U.S. and corresponding foreign regulations, as applicable. We will also be subject to periodic inspections by the FDA and other foreign regulatory authorities as applicable, to determine compliance with U.S. regulatory requirements, such as the Quality System Regulation (QSR), the medical device reporting (MDR), the reporting of adverse events and recalls, the regulations regarding notification on changes and other corresponding regulations of other foreign regulatory authorities, as applicable. These inspections can result in observations or reports, warning letters or other similar notices or forms of enforcement action. If the FDA, or any other foreign authority as applicable, concludes that we are not in compliance with applicable laws or regulations, or that any of our products are ineffective or pose an unreasonable health risk, such authority could ban these products, suspend or cancel our marketing authorizations, impose “stop-sale” and “stop-import” orders, refuse to issue export certificates, detain or seize adulterated or misbranded products, order a recall, repair, replacement, correction or refund of such products, or require us to notify health providers and others that the products present unreasonable risks of substantial harm to the public health. Discovery of previously unknown problems with our product’s design or manufacture may result in restrictions on use, restrictions placed on us or our suppliers, or withdrawal of an existing commercial sale approval. The FDA or comparable foreign authority may also impose operating restrictions, enjoin and restrain certain violations of applicable law pertaining to medical devices, assess civil or criminal penalties against our officers, employees or us, or recommend criminal prosecution of our Company. Adverse regulatory action may restrict us from effectively marketing and selling our products. In addition, negative publicity and product liability claims resulting from any adverse regulatory action could have a material adverse effect on our business, financial condition and operating results.
Foreign governmental regulations have become increasingly stringent and more extensive, and we may become subject to even more rigorous regulation by foreign governmental authorities in the future. Penalties for a company’s non-compliance with foreign governmental regulation could be severe, including revocation or suspension of a company’s business license and civil or criminal sanctions.
Our success depends on our ability to complete development, commercialize, and gain market acceptance initially for PressureSafe and thereafter for any other device.
Our current business strategy is highly dependent on developing and commercially launching one product initially, our PressureSafe device, and achieving and maintaining market acceptance. We may face challenges convincing physicians, many of whom have extensive experience with competitors’ products and established relationships with other companies, to appreciate the benefits of initially PressureSafe in a way that is superior to and differentiated from currently available technology or know-how and adapt it for supporting diagnostics for their patients.
Moreover, healthcare providers tend to be slow to change their medical treatment practices because of perceived liability risks arising from the use of new products and the uncertainty of third-party reimbursement.
If we are unable to achieve the support of caregivers and healthcare providers or widespread market acceptance for our devices, then our sales potential, strategic objectives, and profitability could be negatively impacted, which would adversely affect our business, financial condition, and operating results.
We depend on the knowledge and skills of our senior management and our Board of Directors.
We have benefited substantially from the leadership and performance of our senior management. Our success will depend on our ability to retain our current management and recruit additional management personnel. Competition for senior management in our industry is intense, and we cannot guarantee that we will be able to retain our personnel or recruit additional personnel. The loss of the services of certain members of our senior management could prevent or delay the implementation and completion of our strategic objectives or divert management’s attention to seeking qualified replacements. On February 22, 2024, as a result of financial difficulties, we notified seven of our 10 employees, including Mr. Di-Cori, who acted as our Chief Executive Officer, of the termination of their employment. The effective termination dates vary based on contractual notice periods, which range between March 22, 2024, and April 22, 2024. In addition, our Board of Directors has determined to limit our operations until sufficient funds that can support our operations have been identified.
| 30 |
It may be difficult to enforce a U.S. judgment against us, our officers and directors and the foreign persons named in this registration statement in the United States or in foreign countries, or to assert U.S. securities laws claims in foreign countries or serve process on our officers and directors and these experts.
While we are incorporated in the State of Nevada, currently a majority of our directors and executive officers are not residents of the United States. The majority of our assets are located outside the United States. Therefore, it may be difficult for an investor, or any other person or entity, to enforce a U.S. court judgment based upon the civil liability provisions of the U.S. federal securities laws against us or any of these persons in a U.S. or foreign court, or to effect service of process upon these persons in the United States. Additionally, it may be difficult for an investor, or any other person or entity, to assert U.S. securities law claims in original actions instituted in foreign countries. Foreign courts may refuse to hear a claim based on a violation of U.S. securities laws on the grounds that foreign countries are not necessary the most appropriate forum in which to bring such a claim. Even if a foreign court agrees to hear a claim, it may determine that foreign law and not U.S. law is applicable to the claim. If U.S. law is found to be applicable, the content of applicable U.S. law must be proved as a fact, which can be a time-consuming and costly process. Certain matters of procedure will also be governed by foreign countries’ law’. There is little binding case law in foreign countries addressing the matters described above.
The size and future growth in the market for our planned devices under development has not been established with precision and may be smaller than we estimate, possibly materially. If our estimates and projections overestimate the size of this market, our sales growth may be adversely affected.
Our estimates of the size and future growth in the market for our intended devices under development, including the number of people who may benefit from and be amenable to using our devices for diagnosis, is based on a number of internal and third-party studies, reports and estimates. In addition, our internal estimates are based in large part on current diagnostic patterns by healthcare providers using current generation technology, and our belief is that the incidence of misdiagnosed skin pressure injuries and ear infections in children in the United States and worldwide is increasing. While we believe these factors have historically provided and may continue to provide us with effective tools in estimating the total market for our intended products under development, these estimates may not be correct, and the conditions supporting our estimates may change at any time, thereby reducing the predictive accuracy of these underlying factors. The actual incidence of these phenomenon, and the actual demand for our products or competitive products, could differ materially from our projections if our assumptions are incorrect. As a result, our estimates of the size and future growth in the market for our intended products may prove to be incorrect, it may impair our projected sales growth and have an adverse impact on our business.
Undetected errors or defects in our planned medical devices under development or future versions thereof could harm our reputation, decrease the market acceptance of PressureSafe and DiaDafe.
The technology underlying PressureSafe and Diasafe may contain undetected errors or defects. Disruptions or other performance problems with devices may delay development, prevent regulatory clearance or harm our reputation. If that occurs, we may incur significant costs, the attention of our key personnel could be diverted, or other significant customer relations problems may arise. We may also be subject to warranty and liability claims for damages related to errors or defects in the PressureSafe and Diasafe devices or future versions thereof. A material liability claims or other occurrence that harms our reputation or decreases market acceptance of our planned products could harm our business and operating results. This risk exists even if a device is available for commercial sale and manufactured.
Any product candidates we may advance into clinical trials (assuming the FDA so requires) may be subject to extensive regulation, which can be costly and time-consuming, cause unanticipated delays or prevent the receipt of the required approvals to commercialize our product candidates, all of which can adversely affect our business.
Before we can market a new medical device, such as our proposed products, we must first receive clearance under Section 510(k) of the FDA. In the 510(k) clearance process, before a device may be marketed in the US, the FDA must determine that such proposed device is “substantially equivalent” to a legally marketed “predicate” device, which includes a device that has been previously cleared through the 510(k) process, a device that was legally marketed before May 28, 1976 (pre-amendments device), a device that was originally on the U.S. market under an approved pre-market approval (“PMA”) and later down-classified, or a 510(k)-exempt device. To be “substantially equivalent,” the proposed device must have the same intended use as the predicate device and either have the same technological characteristics as the predicate device or have different technological characteristics and not raise different questions of safety or effectiveness than the predicate device.
| 31 |
The 510(k)-clearance process can be expensive, lengthy, and uncertain. The FDA’s 510(k) clearance process usually takes from three to 12 months but can last longer. Despite the time, effort and cost, a device may not be cleared by the FDA. Any delay or failure to obtain necessary regulatory clearances could harm our business, including our ability to commercialize our product and our shareholders could lose their entire investment. Furthermore, even if we are granted the required regulatory clearances, such clearances may be subject to significant limitations on the indicated uses for the device, which may limit the market for our product.
As noted, on April 9, 2024, the PressureSafe decision support device received an FDA listing certification. PressureSafe is classified as a Class I device. We are currently working on completing larger scale production of the commercial version of the PressureSafe device, planned to be launched during September 2024, following the listing under the FDA. However, no assurance can be granted that we will receive 510(K) clearances for our additional products. If the 510(k) clearance is not granted to us, the device testing, clinical trials, manufacturing, labeling, storage, record-keeping, advertising, promotion, import, export, marketing and distribution of our additional product candidates are subject to extensive regulation by the FDA in the United States and by comparable health authorities in foreign markets.
Despite the time and expense invested in clinical trials of product candidates, commercial sale approval from applicable regulatory authorities is never guaranteed.
FDA or other regulatory agencies can delay, limit, or deny approval of a product candidate for many reasons, including:
| ● | the FDA or other foreign regulatory authority as applicable may disagree with the design or implementation of our clinical trials; | |
| ● | we may be unable to demonstrate to the satisfaction of the FDA that a product candidate is safe and effective for any indication; | |
| ● | the FDA may not accept the clinical data from trials which are conducted by individual investigators in countries where the standard of care is potentially different from the United States; | |
| ● | the results of clinical trials may not meet the level of statistical significance required by the FDA for clearance; | |
| ● | the FDA may disagree with our interpretation of data from the bench testing or clinical trials; | |
| ● | the FDA may fail to approve the manufacturing processes or facilities of third-party manufacturers with which we or our collaborators contract for clinical and commercial supplies; or | |
| ● | the approval policies or regulations of the FDA may significantly be changed in a manner rendering our clinical data insufficient for approval. |
In addition, the FDA may change its clearance and approval policies, adopt additional regulations or revise existing regulations, or take other actions, that may prevent or delay approval or clearance of our products or impact our ability to modify our products after clearance on a timely basis. Such policy or regulatory changes could impose additional requirements upon us that could delay our ability to obtain clearance for our devices, increase the costs of compliance or restrict our ability to maintain products after clearance. For example, as part of the Food and Drug Administration Safety and Innovation Act, or FDASIA, enacted in 2012, Congress reauthorized the Medical Device User Fee Amendments with various FDA performance goal commitments and enacted several “Medical Device Regulatory Improvements” and miscellaneous reforms, which are further intended to clarify and improve medical device regulation both pre- and post-clearance. Some of these proposals and reforms could impose additional regulatory requirements upon us that could delay our ability to obtain new clearance, increase the costs of compliance, or restrict our ability to maintain any commercial sale approval we can obtain.
| 32 |
Concerning foreign markets, approval procedures vary among countries and can involve additional product testing and administrative review periods. Any delay in obtaining, or an inability to obtain, applicable regulatory approvals would prevent us from commercializing our product candidates.
Significant disruptions of information technology systems or security breaches could adversely affect our business.
We are increasingly dependent upon information technology systems, infrastructure and data to operate our business. In the ordinary course of business, we collect, store and transmit large amounts of confidential information (including, among other things, trade secrets or other intellectual property, and proprietary business information). We must do so in a secure manner to maintain the confidentiality and integrity of such confidential information. We also have outsourced elements of our operations to third parties, and as a result, we manage several third-party vendors who may or could have access to our confidential information. The size and complexity of our information technology systems, and those of third-party vendors with whom we contract, and the large amounts of confidential information stored on those systems, make such systems vulnerable to service interruptions or to security breaches from inadvertent or intentional actions by our employees, third-party vendors and/or business partners, or to cyber-attacks by malicious third parties. Cyber-attacks are increasing in frequency, sophistication, and intensity, and have become increasingly difficult to detect. Cyber-attacks could include the deployment of harmful malware, ransomware, denial-of-service attacks, social engineering and other means to affect service reliability and threaten the confidentiality, integrity and availability of information.
Significant disruptions of our information technology systems, or those of our third-party vendors or business partners, or security breaches could adversely affect our business operations and/or result in the loss, misappropriation and/or unauthorized access, use or disclosure of, or the prevention of access to, confidential information, including, among other things, trade secrets or other intellectual property, and proprietary business information, and could result in financial, legal, business and reputational harm to us. Security breaches and other inappropriate access can be difficult to detect, and any delay in identifying them may lead to increased harm of the type described above. While we have implemented security measures to protect our information technology systems and infrastructure, there can be no assurance that such measures will prevent service interruptions or security breaches that could adversely affect our business. In addition, our liability insurance may not be sufficient in type or amount to cover us against costs of or claims related to security breaches, cyber-attacks, and other related breaches. A cybersecurity breach could adversely affect our reputation and could result in other negative consequences, including disruption of our internal operations, increased cybersecurity protection costs, lost revenue, or litigation.
We may be subject to numerous and varying privacy and security laws, and our failure to comply could result in penalties and reputational damage.
We are subject to laws and regulations covering data privacy and the protection of personal information, including health information. The legislative and regulatory landscape for privacy and data protection continues to evolve, and there has been an increasing focus on privacy and data protection issues which may affect our business. In the U.S., numerous federal and state laws and regulations, including state security breach notification laws, state health information privacy laws, and federal and state consumer protection laws, govern the collection, use, disclosure, and protection of personal information. Each of these laws is subject to varying interpretations by courts and government agencies, creating complex compliance issues for us. If we fail to comply with applicable laws and regulations, we could be subject to penalties or sanctions, including criminal penalties if we knowingly obtain or disclose individually identifiable health information from a covered entity in a manner that is not authorized or permitted by the HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act.
| 33 |
Other countries have, or are developing, laws governing the collection, use and transmission of personal information as well. The EU and other jurisdictions have adopted data protection laws and regulations, which impose significant compliance obligations. In the EU, for example, effective May 25, 2018, the General Data Protection Regulation (GDPR) replaced the prior EU Data Protection Directive (95/46) that governed the processing of personal data in the European Union. The GDPR imposes significant obligations on controllers and processors of personal data, including, as compared to the prior directive, higher standards for obtaining consent from individuals to process their personal data, more robust notification requirements to individuals about the processing of their personal data, a strengthened individual data rights regime, mandatory data breach notifications, limitations on the retention of personal data and increased requirements pertaining to health data, and strict rules and restrictions on the transfer of personal data outside of the EU, including to the U.S. The GDPR also imposes additional obligations on, and required contractual provisions to be included in, contracts between companies subject to the GDPR and their third-party processors that relate to the processing of personal data. The GDPR allows EU member states to make additional laws and regulations further limiting the processing of genetic, biometric or health data.
Any failure to comply with the requirements of GDPR and applicable national data protection laws of EU member states could lead to regulatory enforcement actions and significant administrative and/or financial penalties against us (including fines of up to €20,000,000 or up to 4% of the total worldwide annual turnover of the preceding financial year, whichever is higher), and could adversely affect our business, financial condition, cash flows and results of operations.
If we or our third-party manufacturers fail to comply with the FDA’s Quality System Regulation or QSR, our manufacturing operations could be interrupted.
In the US, we and our future contract manufacturers are required to comply with the FDA’s QSR requirements which cover the methods and documentation of the design, testing, production, quality control, labeling, packaging, storage shipping and distribution of our products. In other foreign countries, International Organization for Standardization (ISO) 13485 standard is used to show compliance with the design and manufacturing requirements. We and our suppliers are also subject to the regulations of foreign jurisdictions regarding the manufacturing process if we or our distributors market our products abroad. We continue to monitor our quality management in order to improve our overall level of compliance. Our facilities will be subject to periodic and unannounced inspection by U.S. and other foreign regulatory agencies as applicable to audit compliance with the regulations. If our facilities or those of our suppliers are found to be in violation of applicable laws and regulations, or if we or our suppliers fail to take satisfactory corrective action in response to an adverse inspection, the regulatory authority could take enforcement action, including any of the following sanctions:
| ● | untitled letters, warning letters, fines, injunctions, consent decrees and civil penalties; | |
| ● | customer notifications or repair, replacement or refunds; | |
| ● | operating restrictions or partial suspension or total shutdown of production; | |
| ● | recalls, withdrawals, or administrative detention or seizure of our products; | |
| ● | refusing or delaying requests for 510(k) marketing clearance applications relating to new products or modified products; | |
| ● | withdrawing the product from the market; | |
| ● | refusing to provide Certificates for Foreign Government; | |
| ● | refusing to grant export approval for our products; or | |
| ● | pursuing criminal prosecution. |
I
Any of these sanctions could impair our ability to produce PressureSafe, DiaSafe or Nobiotics in a cost-effective and timely manner to meet our customers’ demands and could have a material adverse effect on our reputation, business, results of operations, and financial condition. We may also be required to bear other costs or take other actions that may hurt our future sales and our ability to generate profits.
| 34 |
We intend to rely on third parties to conduct clinical trials (if needed). If these third parties do not meet our deadlines or otherwise conduct the trials as required, our clinical trials programs could be delayed or unsuccessful and we may not be able to obtain regulatory approval for or commercialize our product candidates when expected or at all.
We do not have the ability to conduct all aspects of our clinical trials ourselves. We intend to use Contract Research Organizations (CROs) to conduct clinical trials that we may be required to conduct and will rely upon medical institutions, clinical investigators and CRO’s and consultants to conduct these trials in accordance with our clinical protocols. Our future CROs, investigators and other third parties play a significant role in the conduct of these trials and the subsequent collection and analysis of data from the clinical trials.
There is no guarantee that any CROs, investigators and other third parties upon which we rely for administration and conduct of clinical trials will devote adequate time and resources to such trials or perform as contractually required. If any of these third parties fail to meet expected deadlines, fail to adhere to our clinical protocols or otherwise perform in a substandard manner, our clinical trials may be extended, delayed or terminated. If any of these clinical trial sites terminate for any reason, we may experience the loss of follow-up information on patients enrolled in our ongoing clinical trials unless we are able to transfer the care of those patients to another qualified clinical trial site. In addition, principal investigators for any clinical trials we conduct may serve as scientific advisors or consultants to us from time to time and receive cash or equity compensation in connection with such services. If these relationships and any related compensation result in perceived or actual conflicts of interest, the integrity of the data generated at the applicable clinical trial site may be jeopardized.
If our competitors develop tools for the target indications of our product candidates that are approved more quickly, marketed more successfully or demonstrated to be more effective or accurate than our product candidates, our commercial opportunity will be reduced or eliminated.
We operate in highly competitive segments of the medical device markets. We face competition from many different sources, including commercial medical device enterprises, academic institutions, government agencies and private and public research institutions. Our product candidates, if successfully developed and approved, will compete with established methods, as well as new diagnostic technologies that may be introduced by our competitors. Our competitors may have significantly greater financial, product development, manufacturing and marketing resources than us. Large medical device companies have extensive experience in clinical testing and obtaining regulatory approval for medical devices. We also may compete with these organizations to recruit management, scientists and clinical development personnel. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. New developments, including the development of other medical device technologies and methods of pressure injuries and ear infections diagnostics, may occur in the medical device industries at a rapid pace. Developments by competitors may render our product candidates obsolete or non-competitive. We will also face competition from these third parties in recruiting and retaining qualified personnel, establishing clinical trial sites and patient registration for clinical trials and in identifying and in-licensing new product candidates.
If we or our U.S. Distributor are unable to establish sales and marketing capabilities or fail to enter into agreements with third parties to market and sell any products we may successfully develop, we may not be able to effectively market and sell any such products and generate product revenue.
In October 2022, we entered into a distribution agreement covering the U.S. pursuant to which, among other things, the distributor is to assist us in setting up a distribution network in the U.S. The establishment and development of a sales force, either by us or jointly with distributor, or the establishment of a contract sales force to market any products we may develop will be expensive and time-consuming and could delay any product launch. If we, or our development partners, are unable to establish sales and marketing capability or any other non-technical capabilities necessary to commercialize any products we may successfully develop, we will need to contract with third parties to market and sell such products. We may not be able to establish arrangements with third parties on acceptable terms, if at all.
| 35 |
If we are not able to develop a strong brand and/ or increase market awareness for our product candidates, then our business, results of operations and financial condition may be adversely affected.
We believe that the success of our product candidates will depend in part on our ability to develop a strong brand identity for our company and products, and to increase the market awareness of our product and their capabilities, once these products are commercially launched. The successful promotion of our brand will depend largely on our continued marketing efforts and our ability to offer high quality AI capabilities with our products and ensure that our technology provides the expected benefits. Our brand promotion and thought leadership activities may not be successful or produce revenue. In addition, independent industry analysts may provide reviews of our products and of competing products and services, which may significantly influence the perception of our products in the marketplace. If these reviews are negative or not as positive as reviews of our competitors’ products and services, then our brand may be harmed.
The promotion of our brand also requires us to make substantial expenditures, and we anticipate that these expenditures will increase as our industry becomes more competitive and as we seek to expand into new markets. These higher expenditures may not result in any increased revenue or in revenue that is sufficient to offset the higher expense levels. If we do not successfully maintain and enhance our brand, then our business may not grow, we may see our pricing power reduced relative to competitors and we may lose customers, all of which would adversely affect our business, results of operations and financial condition.
Failure to manage our growth effectively could increase our expenses, decrease our revenue and prevent us from implementing our business strategy.
We expect that our ability to generate revenues and achieve profitability will require substantial growth in our business, which will put a strain on our management and financial resources. To manage this and our anticipated future growth effectively, including as we expand into new clinical areas and geographic regions, we must continue to maintain and enhance our information technology infrastructure, as well as our financial and accounting systems and controls. We also must attract, train and retain a significant number of qualified software and hardware developers and engineers, technical and management personnel, sales and marketing personnel and customer and channel partner support personnel. Failure to effectively manage our rapid growth could lead us to over-invest or under-invest in development and operations, result in weaknesses in our systems or controls, give rise to operational mistakes, losses, loss of productivity or business opportunities and result in loss of employees and reduced productivity of remaining employees. Our growth could require significant capital expenditures and might divert financial resources from other projects such as the development of new products and services. If our management is unable to effectively manage our growth, our expenses might increase more than expected, our revenue could decline or grow more slowly than expected, and we might be unable to implement our business strategy. The quality of our products and services might suffer, which could negatively affect our reputation and harm our ability to retain and attract channel partners or customers.
Failure to secure or retain coverage or adequate reimbursement for our planned products in development by third-party payors could adversely affect our business, financial condition and operating results.
We plan to derive nearly all of our revenue from sales, initially, of our PressureSafe device under development, if approved for commercial sale, in the United States and potentially in selected international geographies and expect to do so for the next several years. We anticipate a substantial portion of the purchase price of our product and disposables will be paid for by third-party payors, including private insurance companies, preferred provider organizations, and other managed care providers. Patients who receive services for their medical conditions and their healthcare providers generally rely on third-party payors to reimburse all or part of the costs associated with their medical treatment and diagnosis, including healthcare providers’ services. Coverage and adequate reimbursement from third-party payors, including governmental healthcare programs, such as Medicare and Medicaid, and commercial payors, is critical to new product acceptance. Future sales of our PressureSafe device initially will be limited unless healthcare providers can rely on third-party payors to pay for all or part of the cost to purchase/lease our devices and then pay for the disposable components. Access to adequate coverage and reimbursement by third-party payors is essential to the market acceptance of our products.
| 36 |
In the United States, a third-party payor’s decision to provide coverage for our products does not imply that an adequate reimbursement rate will be obtained. Further, one third-party payor’s decision to cover our products does not assure that other payors will also provide coverage for the products or will provide coverage at an adequate reimbursement rate. Healthcare providers may choose not to order a product and/or disposables unless third-party payors pay a substantial portion of the product and disposables. Within and outside the United States, reimbursement is obtained from a variety of sources, including government-sponsored and private health insurance plans. These third-party payors determine whether to provide coverage and reimbursement for specific products and procedures. Coverage determinations and reimbursement levels of our products are critical to the commercial success of our product, and if we are not able to secure positive coverage determinations and reimbursement levels for our products, our business would be materially adversely affected.
In addition, there may be significant delays in obtaining reimbursement, and coverage may be more limited than the purposes for which the product received commercial sale approval from the FDA or other foreign regulatory authorities. Moreover, eligibility for reimbursement does not imply that any product will be paid for in all cases or at a rate that covers our costs, including research, development, manufacture, sale and distribution. Payment rates may vary according to the use of the product and the clinical setting in which it is used, may be based on payments allowed for lower-cost products that are already reimbursed, and may be incorporated into existing payments for other services. Net prices for products may be reduced by mandatory discounts or rebates required by government healthcare programs or third-party payors and by any future relaxation of laws that presently restrict imports of products from countries where they may be sold at lower prices than in the United States.
Third-party payors, whether foreign or domestic, governmental or commercial, are developing increasingly sophisticated methods of controlling healthcare costs. In addition, in the United States, no uniform policy of coverage and reimbursement for medical device products and services exists among third-party payors. Therefore, coverage and reimbursement for medical device products and services can differ significantly from payor to payor. In addition, payors continually review new technologies for possible coverage and can, without notice, deny coverage for these new products and procedures. As a result, the coverage determination process is often a time-consuming and costly process that will require us to provide scientific and clinical support for the use of our products to each payor separately, with no assurance that coverage and adequate reimbursement will be obtained or maintained if obtained.
Reimbursement systems in international markets vary significantly by country and by region within some countries, and reimbursement approvals must be obtained on a country-by-country basis. In many international markets, a product must be approved for reimbursement before it can be approved for sale in that country. Further, many international markets have government-managed healthcare systems that control reimbursement for new devices and procedures. In most markets, there are private insurance systems as well as government-managed systems. If sufficient coverage and reimbursement are not available for any product we develop, in either the United States or internationally, the demand for our products and our revenues will be adversely affected.
If we fail to attract and retain key management and R&D personnel, we may be unable to successfully develop or commercialize our product candidates.
As stated above, on February 22, 2024, as a result of financial difficulties, we notified seven of our 10 employees of the termination of their employment. We will need to expand and effectively manage our managerial, operational, financial, and other resources to successfully pursue our product development and commercialization efforts. As a company with a limited number of personnel, we are highly dependent on the development, regulatory, commercial, and financial expertise of the members of our senior management. The loss of such individuals or the services of any of our other senior management could delay or prevent the further development and potential commercialization of our product candidates and, if we are not successful in finding suitable replacements, could harm our business. Our success also depends on our continued ability to attract, retain and motivate highly qualified management and scientific personnel and we may not be able to do so in the future due to the intense competition for qualified personnel among biotechnology, medical device and high-technology and companies, as well as universities and research organizations. If we are not able to attract and retain the necessary personnel, we may experience significant impediments to our ability to implement our business strategy.
| 37 |
We may seek to grow our business through acquisitions of complementary products or technologies, and the failure to manage acquisitions, or the failure to integrate them with our existing business, could have a material adverse effect on our business, financial condition and operating results.
From time to time, we may consider opportunities to acquire other products or technologies that may enhance our products, platform or technology, expand the breadth of our markets or customer base, or advance our business strategies. Potential acquisitions involve numerous risks, including:
| ● | problems assimilating the acquired products or technologies; | |
| ● | issues maintaining uniform standards, procedures, controls and policies; | |
| ● | unanticipated costs associated with acquisitions; | |
| ● | diversion of management’s attention from our existing business; | |
| ● | risks associated with entering new markets in which we have limited or no experience; and | |
| ● | increased legal and accounting costs relating to the acquisitions or compliance with regulatory matters. |
We have no current commitments with respect to any acquisition. We do not know if we will be able to identify suitable acquisitions, whether we will be able to successfully complete any such acquisitions on favorable terms or at all, or whether we will be able to successfully integrate any acquired products or technologies. Our potential inability to integrate any acquired products or technologies effectively may adversely affect our business, operating results and financial condition.
Risks Related to our Intellectual Property
If we are unable to protect our intellectual property rights or if our intellectual property rights are inadequate to protect our technology, our competitors could develop and commercialize technology similar to ours, and our competitive position could be harmed.
We rely on a combination of patent and trademark laws in the United States and other countries, trade secret protection, confidentiality agreements and other contractual arrangements with our employees, consultants and others to maintain our competitive position. In particular, our success depends, in part, on our ability to maintain patent protection for our products, technologies and inventions, maintain the confidentiality of our trade secrets and know-how, operate without infringing upon the proprietary rights of others and prevent others from infringing upon our proprietary rights. Despite our efforts to protect our proprietary rights, it is possible that competitors or other unauthorized third parties may obtain, copy, use or disclose our technologies, inventions, processes or improvements. Moreover, other parties may independently develop similar or competing technology, methods, know-how or design around any patents that may be issued to or held by us. Unauthorized parties may also attempt to copy or reverse engineer certain aspects of our products that we consider proprietary. We cannot assure you that our existing or any future patents or other intellectual property rights will not be challenged, invalidated or circumvented, or will otherwise provide us with meaningful protection. If our patents and other intellectual property do not adequately protect our technology, our competitors may be able to offer products similar to ours. Our competitors may also be able to develop similar technology independently or design around any patent(s) granted to us, and we may not be able to detect the unauthorized use of our proprietary technology or take appropriate steps to prevent such use.
Any such activities by our competitors that circumvent our intellectual property protection could subvert our competitive advantage and have an adverse effect on our results of operations.
| 38 |
Furthermore, filing, prosecuting, maintaining and defending patents on our solutions in all countries throughout the world would be prohibitively expensive, and our intellectual property rights in some countries outside the United States are less extensive than those in the United States. In addition, the laws of some foreign countries do not protect intellectual property rights to the same extent as federal and state laws in the United States. Also, it may not be possible to effectively enforce intellectual property rights in some foreign countries at all or to the same extent as in the United States and other countries. Consequently, we may be unable to prevent third parties from using our inventions in all countries, or from selling or importing products made using our inventions in the jurisdictions in which we do not have (or are unable to effectively enforce) patent protection. Competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop, market or otherwise commercialize their own products, and we may be unable to prevent those competitors from importing those infringing products into territories where we have patent protection, but enforcement is not as strong as in the United States.
We may be sued by third parties for alleged infringement of their proprietary rights, which could adversely affect our business, results of operations and financial condition.
There is often litigation between competing companies relying on their respective technologies based on allegations of infringement or other violations of intellectual property rights. Our future success depends, in part, on not infringing the intellectual property rights of others. We may be unaware of the intellectual property rights of others that may cover some or all of our technology. Any such claims or litigation could cause us to incur significant expenses and, if successfully asserted against us, could require that we pay substantial damages or ongoing royalty payments, prevent us from offering some portion of our products, or require that we comply with other unfavorable terms. We may also be obligated to indemnify our customers or channel partners in connection with any such litigation and to obtain licenses or modify our products, which could further exhaust our resources. Patent infringement, trademark infringement, trade secret misappropriation, and other intellectual property claims and proceedings brought against us, whether successful or not, could harm our brand, business, results of operations and financial condition. Litigation is inherently uncertain, and any judgment or injunctive relief entered against us, or any adverse settlement could negatively affect our business, results of operations and financial condition. In addition, litigation can involve significant management time and attention and be expensive, regardless of the outcome. During litigation, there may be announcements of the results of hearings and motions and other interim developments related to the litigation. If securities analysts or investors regard these announcements as negative, the trading price of our ordinary shares may decline.
We may become involved in lawsuits to protect or enforce our patents, which could be expensive, time-consuming and ultimately unsuccessful.
If we attempt enforcement of our patents or other intellectual property rights, we may be subject or party to claims, negotiations, or complex, protracted litigation. These claims and any resulting lawsuits, if resolved adversely to us, could subject us to significant liability for damages, impose temporary or permanent injunctions against our solutions or business operations, or invalidate or render unenforceable our intellectual property.
Intellectual property disputes and litigation, regardless of merit, can be costly and disruptive to our business operations by diverting the attention and energies of management and key technical personnel, and by increasing our costs of doing business. Such litigation, regardless of its success, could seriously harm our reputation with our channel partners, business partners, and patients and in the industry at large. Some of our competitors may be able to sustain the costs of complex patent or intellectual property litigation more effectively than we can because they have substantially greater resources. Any of the foregoing could adversely affect our operating results.
Risks Relating to Our Israel Operations
Our technology development personnel are headquartered in Israel and, therefore, our results may be adversely affected by economic restrictions imposed on, and political and military instability in, Israel.
Our technology development headquarters, which houses substantially all of our research and development team, including engineers, machinists, researchers, and clinical and regulatory personnel as well as the facility of our contract manufacturer and final assembly are located in Israel. Our employees, service providers, directors and officers are residents of Israel. Accordingly, political, economic and military conditions in Israel and the surrounding region may directly affect our business. Any hostilities involving Israel or the interruption or curtailment of trade within Israel or between Israel and its trading partners could materially and adversely affect our business, financial condition and results of operations and could make it more difficult for us to raise capital. Although we plan to maintain inventory in the United States and Germany, an extended interruption could materially and adversely affect our business, financial condition and results of operations.
| 39 |
In October 2023, Hamas terrorists infiltrated Israel’s southern border from the Gaza Strip and conducted a series of attacks on civilian and military targets. Hamas also launched extensive rocket attacks on Israeli population and industrial centers located along Israel’s border with the Gaza Strip and in other areas within the State of Israel. These attacks resulted in extensive deaths, injuries and kidnapping of civilians and soldiers. Following the attack, Israel’s security cabinet declared war against Hamas, and a military campaign against these terrorist organizations commenced in parallel to their continued rocket and terror attacks.
The intensity and of Israel’s current war against Hamas is difficult to predict, as are such war’s economic implications on the Company’s business and operations and on Israel’s economy in general. These events may be intertwined with wider macroeconomic indications of a deterioration of Israel’s economic standing, which may have a material adverse effect on the Company and its ability to effectively conduct some of its operations.
Our commercial insurance does not cover losses that may occur as a result of events associated with war and terrorism. Following the attack by Hamas on Israel’s southern border, Hezbollah in Lebanon has also launched missile, rocket and shooting attacks against Israeli military sites, troops, and Israeli towns in northern Israel. In response to these attacks, the Israeli army has carried out a number of targeted strikes on sites belonging to Hezbollah in southern Lebanon. It is possible that other terrorist organizations, including Palestinian military organizations in the West Bank, as well as other hostile countries, such as Iran, will join the hostilities. Such hostilities may include terror and missile attacks. Any hostilities involving Israel or the interruption or curtailment of trade between Israel and its trading partners could adversely affect our operations and results of operations. Any armed conflicts, terrorist activities or political instability in the region could materially and adversely affect our business, financial condition and results of operations.
Our operations and the operations of our contract manufacturer may be disrupted as a result of the obligation of Israeli citizens to perform military service.
Many Israeli citizens are obligated to perform one month, and in some cases more, of annual military reserve duty until they reach the age of 45 (or older, for reservists with certain occupations) and may be called to active duty. In connection with the Israeli security cabinet’s declaration of war against Hamas and possible hostilities with other organizations, several hundred thousand Israeli military reservists were drafted to perform immediate military service. One of our employees and consultants (and their spouses or partners) in Israel have been called, and additional employees (or their spouses or partners) may be called, for service in the current or future wars or other armed conflicts with Hamas, and such persons may be absent for an extended period of time. As a result, our operations in Israel may be disrupted by such absences, which disruption may materially and adversely affect our business, prospects, financial condition and results of operations.
Our sales may be adversely affected by boycotts of Israel.
Several countries, principally in the Middle East, restrict doing business with Israel and Israeli companies, and additional countries may impose restrictions on doing business with Israel and Israeli companies whether as a result of hostilities in the region or otherwise, and specifically following the Israel- Hamas war. In addition, there have been increased efforts by activists to cause companies and consumers to boycott Israeli goods based on Israeli government policies. Such actions, particularly if they become more widespread, may adversely impact our ability to sell our products.
| 40 |
Our subsidiary has received Israeli government grants for certain of our research and development activities and we may receive additional grants in the future. The terms of those grants restrict our ability to transfer technologies outside of Israel, and we may be required to pay penalties in such cases or upon the sale of our company.
Our subsidiary, IR. Med Ltd., received a total of $509,000 from the Israel Innovation Authority (IIA). We may in the future apply to receive additional grants from the IIA to support our research and development activities. With respect to such grants, we are committed to pay royalties at a rate of 3.0% to 3.5% on sales proceeds up to the total amount of grants received, linked to the U.S. dollar and bearing interest at an annual rate of LIBOR applicable to dollar deposits. Until October 25, 2023, the interest was calculated at a rate based on 12-month LIBOR applicable to U.S. Dollar deposits. However, on October 25, 2023, the IIA published a directive concerning changes in royalties to address the expiration of the LIBOR. Under such directive, regarding IIA grants approved by the IIA prior to January 1, 2024 but which are outstanding thereafter, as of January 1, 2024 the annual interest is calculated at a rate based on 12-month SOFR, or at an alternative rate published by the Bank of Israel plus 0.71513%; and, for grants approved on or following January 1, 2024, the annual interest shall be the higher of (i) the 12 months SOFR interest rate, plus 1%, or (ii) a fixed annual interest rate of 4%. Even after payment in full of these amounts, we will still be required to comply with the requirements of the Israeli Encouragement of Industrial Research, Development and Technological Innovation Law, 1984, or the R&D Law, and related regulations, with respect to those past grants. When a company develops know-how, technology or products using IIA grants, the terms of these grants and the R&D Law restrict the transfer outside of Israel of such know-how, and of the manufacturing or manufacturing rights of such products, technologies or know-how, without the prior approval of the IIA. Therefore, if aspects of our technologies are deemed to have been developed with IIA funding, the discretionary approval of an IIA committee would be required for any transfer to third parties outside of Israel of know-how or manufacturing or manufacturing rights related to those aspects of such technologies. Furthermore, the IIA may impose certain conditions on any arrangement under which it permits us to transfer technology or development out of Israel or may not grant such approvals at all.
Furthermore, the consideration available to our shareholders in a future transaction involving the transfer outside of Israel of technology or know-how developed with IIA funding (such as a merger or similar transaction) may be reduced by any amounts that we are required to pay to the IIA. Any such mergers require IIA approval to avoid penalties.
In addition to the above, any non-Israeli citizen, resident or entity that, among other things, (i) becomes a holder of 5% or more of our share capital or voting rights, (ii) is entitled to appoint one or more of our directors or our chief executive officer or (iii) serves as one of our directors or as our chief executive officer (including holders of 25% or more of the voting power, equity or the right to nominate directors in such direct holder, if applicable) is required to notify the IIA and undertake to comply with the rules and regulations applicable to the grant programs of the IIA, including the restrictions on transfer described above. Such notification will be required in connection with the investment being made by an investor.
Risks Related to the Ownership of our Common Stock and This Offering
There is not now, and there may never be, an active, liquid and orderly trading market for our common stock, which may make it difficult for you to sell your shares of our common stock.
There is not now, nor has there been since our inception, an orderly and liquid market for shares of our common stock, and an active trading market for our shares may never develop or be sustained after this offering. As a result, investors in our common stock must bear the economic risk of holding those shares for an indefinite period of time. Our common stock is quoted on the OTCQB-tier of the OTC Markets, an over-the-counter quotation system. An active market for our common stock may never develop or be sustained. If an active market for our common stock does not develop, it may be difficult for you to sell the shares you purchase in this offering without depressing the market price for the shares or at all. Further, an inactive market may also impair our ability to raise capital by selling additional equity in the future and may impair our ability to enter into strategic partnerships or acquire companies or products by using shares of our common stock as consideration.
Directors, executive officers, principal stockholders and affiliated entities own a significant percentage of our capital stock, and they may make decisions that our stockholders do not consider to be in their best interests.
Currently, our directors, executive officers, principal stockholders and affiliated entities beneficially own, in the aggregate, approximately 50% of our outstanding voting securities. This concentration of ownership may have the effect of delaying or preventing a change in control of our company that may be favored by other stockholders. This could prevent transactions in which stockholders might otherwise recover a premium for their shares over current market prices. This concentration of ownership and influence in management and board decision-making could also harm the price of our capital stock by, among other things, discouraging a potential acquirer from seeking to acquire shares of our capital stock (whether by making a tender offer or otherwise) or otherwise attempting to obtain control of our company.
| 41 |
Sale of our common stock by our stockholders could encourage short sales by third parties, which could contribute to the further decline of our stock price.
The significant downward pressure on the price of our common stock caused by the sale of material amounts of common stock could encourage short sales by third parties. Such an event could place further downward pressure on the price of our common stock.
There is a limited existing market for our common stock.
Prior to this offering, there has been a limited public market for our common stock. We cannot assure you that a more active trading market for our common stock or warrants will develop following this offering, or if it does develop, that it will be maintained. You may not be able to sell your securities quickly or at the market price if trading in our securities is not active. The public offering price for the securities will be determined by negotiations between us and the representatives of the underwriters and may not be indicative of prices that will prevail in the trading market. Upon closing of this offering, our common stock and warrants will be listed on Nasdaq, however, we cannot ensure that an active public market for our common stock and warrants will develop after this offering, or that if it does develop, it will be sustained. In the absence of an active public trading market:
| ● | you may not be able to resell your securities at or above the public offering price; | |
| ● | the market price of our common stock may experience more price volatility; and | |
| ● | there may be less efficiency in carrying out your purchase and sale orders. |
Our share price is expected to be volatile and may be influenced by numerous factors, some of which are beyond our control.
Market prices for shares of biotechnology and medical device companies such as ours are often volatile, and the quoted price of our common stock is therefore likely to be highly volatile and could be subject to wide fluctuations in response to various factors, some of which are beyond our control. In addition to the factors discussed in this “Risk Factors” section and elsewhere in this report, these factors include:
| ● | the product candidates we seek to pursue and our ability to obtain rights to develop, commercialize and market those candidates; | |
| ● | our decision to initiate a clinical trial, not to initiate a clinical trial or to terminate an existing clinical trial; | |
| ● | actual or anticipated adverse results or delays in our clinical trials; | |
| ● | our failure to commercialize our product candidates, if approved; | |
| ● | unanticipated serious safety concerns related to the use of any of our product candidates; | |
| ● | adverse regulatory decisions; | |
| ● | additions or departures of key scientific or management personnel; | |
| ● | changes in laws or regulations applicable to our product candidates, including without limitation clinical trial requirements for approvals; | |
| ● | disputes or other developments relating to patents and other proprietary rights and our ability to obtain patent protection for our product candidates; | |
| ● | actual or anticipated variations in quarterly operating results; | |
| ● | failure to meet or exceed the estimates and projections of the investment community; |
| 42 |
| ● | overall performance of the equity markets and other factors that may be unrelated to our operating performance or the operating performance of our competitors, including changes in market valuations of similar companies; | |
| ● | conditions or trends in the biotechnology and medical device industries; | |
| ● | introduction of new products offered by us or our competitors; | |
| ● | announcements of significant acquisitions, strategic partnerships, joint ventures or capital commitments by us or our competitors; | |
| ● | our ability to maintain an adequate rate of growth and manage such growth; | |
| ● | issuances of debt or equity securities; | |
| ● | sales of our common stock by us or our stockholders in the future, or the perception that such sales could occur; | |
| ● | trading volume of our common stock; | |
| ● | ineffectiveness of our internal control over financial reporting or disclosure controls and procedures; | |
| ● | general political and economic conditions; | |
| ● | effects of natural or man-made catastrophic events; and | |
| ● | other events or factors, many of which are beyond our control. |
In addition, the stock market in general, and the stocks of small-cap biotechnology and medical device companies in particular, have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of these companies. Broad market and industry factors may negatively affect the market price of our common stock, regardless of our actual operating performance. In addition, other biotechnology and medical device companies or our competitors’ programs could have positive or negative results that impact their stock prices, and their results or stock fluctuations could have a positive or negative impact on our stock price regardless of whether such impact is direct or not. The realization of any of the above risks or any of a broad range of other risks, including those described in these “Risk Factors,” could have a dramatic and material adverse impact on the market price of our common stock.
If securities or industry analysts do not publish, or cease publishing, research or publish inaccurate or unfavorable research about our business or our market, or if they change their recommendations regarding our stock adversely, our stock price and any trading volume could decline.
Any trading market for our common stock that may develop will depend in part on the research and reports that securities or industry analysts publish about us or our business, markets or competitors. Securities and industry analysts do not currently, and may never, publish research on us or our business. If no securities or industry analysts commence coverage of our company, the trading price for our stock would be negatively affected. If securities or industry analysts initiate coverage, and one or more of those analysts downgrade our stock or publish inaccurate or unfavorable research about our business or our market, our stock price would likely decline. If one or more of these analysts cease coverage of our company or fail to publish reports on us regularly, demand for our stock could decrease, which might cause our stock price and any trading volume to decline.
We may have become exposed to material liabilities that were not discovered before, and have not been discovered since, due to the closing of the Acquisition.
As a result of the Acquisition, we are responsible for any liabilities incurred by IR. Med Ltd. IR. Med Ltd. may have material liabilities that have not been discovered or asserted. We could experience losses as a result of any such undisclosed liabilities that are discovered in the future, which could materially harm our business and financial condition. As a result, our current and future stockholders will bear some, or all, of the risks relating to any such unknown or undisclosed liabilities, if any.
| 43 |
We are exposed to additional risks as a result of “going public” by means of a reverse acquisition transaction.
We are exposed to additional risks because the business of IR. Med Ltd. has become a public company through a “reverse acquisition” transaction, or the Acquisition. There has been increased focus in recent years by government agencies on transactions such as the Acquisition, and we may be subject to increased scrutiny by the SEC or other government agencies and holders of our securities as a result of the completion of that transaction. Further, as a result of our existence as a “shell company” under applicable rules of the SEC prior to the closing of the Acquisition, we are subject to certain restrictions and limitations for certain specified periods of time relating to potential future issuances of our securities and compliance with applicable SEC rules and regulations. Additionally, our “going public” by means of a reverse acquisition transaction may make it more difficult for us to obtain coverage from securities analysts of major brokerage firms following the Acquisition because there may be little incentive to those brokerage firms to recommend the purchase of our common stock. Further, investment banks may be less likely to agree to underwrite secondary offerings on our behalf than they might if we became a public reporting company by means of an initial public offering (IPO), because they may be less familiar with our company as a result of more limited coverage by analysts and the media, and because we became public at an early stage in our development. The failure to receive research coverage or support in the market for our shares will have an adverse effect on our ability to develop a liquid market for our common stock. The occurrence of any such event could cause our business or stock price to suffer.
If we fail to maintain proper and effective internal controls, our ability to produce accurate and timely financial statements could be impaired, which could harm our operating results, our ability to operate our business and investors’ views of us.
We are required to comply with Section 404 of the Sarbanes-Oxley Act of 2002, as amended, or the Sarbanes- Oxley Act, subject to certain exceptions. Section 404 of the Sarbanes-Oxley Act requires public companies to conduct an annual review and evaluation of their internal controls and to obtain attestations of the effectiveness of internal controls by independent auditors. As a private company, IR-Med Operations was not subject to requirements to establish, and did not establish, internal control over financial reporting and disclosure controls and procedures prior to the Acquisition. Our management team and Board of Directors will need to devote significant efforts to maintaining adequate and effective disclosure controls and procedures and internal control over financial reporting in order to comply with applicable regulations, which may include hiring additional legal, financial reporting and other finance and accounting staff. Additionally, any of our efforts to improve our internal controls and design, implement and maintain an adequate system of disclosure controls may not be successful and will require that we expend significant cash and other resources.
Ensuring that we have adequate internal financial and accounting controls and procedures in place so that we can produce accurate financial statements on a timely basis is a costly and time-consuming effort that will need to be evaluated frequently. Our failure to maintain the effectiveness of our internal controls in accordance with the requirements of the Sarbanes-Oxley Act could have a material adverse effect on the tradability of our common stock, which in turn would negatively impact our business. We could lose investor confidence in the accuracy and completeness of our financial reports, which could have an adverse effect on the price of our common stock. In addition, if our efforts to comply with new or changed laws, regulations, and standards differ from the activities intended by regulatory or governing bodies due to ambiguities related to practice, regulatory authorities may initiate legal proceedings against us, and our business may be harmed.
If material weaknesses or deficiencies in our internal controls exist and go undetected or unremedied, our financial statements could contain material misstatements that, when discovered in the future, could cause us to fail to meet our future reporting obligations and cause the price of our common stock to decline.
| 44 |
Shares of our common stock that have not been registered under federal securities laws are subject to resale restrictions imposed by Rule 144, including those set forth in Rule 144(i) which apply to a former “shell company.”
We were previously deemed a “shell company” under applicable SEC rules and regulations, prior to the reverse merger transaction in which we became a public company, because we had no or nominal operations and either no or nominal assets, assets consisting solely of cash and cash equivalents, or assets consisting of any amount of cash and cash equivalents and nominal other assets. Pursuant to Rule 144 of the Securities Act, sales of the securities of a former shell company, such as us, are not permitted unless at the time of a proposed sale, (i) we are subject to the reporting requirements of Section 13 or 15(d) of the Exchange Act; and (ii) we have filed all reports and other materials required to be filed by Section 13 or 15(d) of the Exchange Act, as applicable, during the preceding 12 months, other than current reports on Form 8-K. Additionally, our previous status as a shell company could also limit our use of our securities to pay for any acquisitions we may seek to pursue in the future. The lack of liquidity of our securities as a result of the inability to sell under Rule 144 for a longer period of time than a non-former shell company could cause the market price of our securities to decline. Additionally, our previous status as a shell company could also limit our use of our securities to pay for any acquisitions we may seek to pursue in the future (although none are currently planned). The lack of liquidity of our securities as a result of the inability to sell under Rule 144 for a longer period of time than a non-former shell company could cause the market price of our securities to decline.
If we issue additional shares of our capital stock in the future, our existing stockholders will be diluted.
Our Amended and Restated Articles of Incorporation authorizes the issuance of up to 600,000,000 shares of our common stock. Possible business and financial uses for our authorized capital stock include, without limitation, equity financing, such as future stock splits, acquiring other companies, businesses or products in exchange for shares of our capital stock, issuing shares of our capital stock to partners or other collaborators in connection with strategic alliances, attracting and retaining employees by the issuance of additional securities under our equity compensation plan, or other transactions and corporate purposes that our Board of Directors deems are in the interests of our company. Additionally, issuances of shares of our capital stock could have the effect of delaying or preventing changes in control or our management. Any future issuances of shares of our capital stock may not be made on favorable terms or at all; they may have rights, preferences and privileges that are superior to those of our common stock and may have an adverse effect on our business or the trading price of our common stock. The issuance of any additional shares of our common stock will reduce the book value per share and may contribute to a reduction in the market price of the outstanding shares of our common stock.
Sales of a substantial number of shares of our common stock in the public market, or the perception that such sales could occur, could cause our stock price to fall.
If our existing stockholders sell, or indicate an intention to sell, substantial amounts of our common stock in the public market, the trading price of our common stock could decline. As of the date of this report, a total of 69,931,056 shares of our common stock are outstanding. Of those shares, approximately 20,442,611 are currently freely tradable, without restriction, in the public market, and 14,230,259 shares are issuable upon exercise of warrants that are registered for resale under the Securities Act. Any sales of those shares or any perception in the market that such sales may occur could cause the trading price of our common stock to decline.
Anti-takeover provisions in our organizational documents could delay or prevent a change of control.
Provisions of our Amended and Restated Articles of Incorporation and Amended and Restated Bylaws may have an anti-takeover effect and may delay, defer or prevent a merger, acquisition, tender offer, takeover attempt or other change of control transaction that a stockholder might consider to be in its interests, including attempts that might result in a premium over the market price for the shares held by our stockholders.
| 45 |
These provisions provide, among other things:
| ● | a classified Board of Directors with staggered three-year terms; | |
| ● | advance notice for nominations of directors by stockholders and for stockholders to include matters to be considered at stockholder meetings; | |
| ● | certain limitations on convening special stockholder meetings and the prohibition of stockholder action by written consent; and | |
| ● | directors may only be removed for cause and only by the affirmative vote of the holders of at least eighty percent (80%) of the voting power of all of the then-outstanding shares of our capital stock entitled to vote at an election of directors, voting together as a single class. |
These anti-takeover provisions, including those noted above, could make it more difficult for a third party to acquire us, even if the third party’s offer may be considered beneficial by many of our stockholders. As a result, our stockholders may be limited in their ability to obtain a premium for their shares. See “Description of Securities.”
Article XI of our Second Amended and Restated Articles of Incorporation designates the Eighth Judicial District Court of Clark County, Nevada as the sole and exclusive forum for certain types of actions and proceedings that may be initiated by our shareholders and therefore may limit our shareholders’ ability to choose a forum for disputes with us or our directors, officers, employees, or agents.
Article XI of our Second Amended and Restated Articles of Incorporation provides that, to the fullest extent permitted by law, and unless we consent to the selection of an alternative forum, the Eighth Judicial District Court of Clark County, Nevada shall be the sole and exclusive forum for (a) any derivative action or proceeding brought on behalf of the Company, (b) any action or proceeding asserting a claim of breach of a fiduciary duty owed by any director or officer of the Company to the Company or the Company’s shareholders, (c) any action or proceeding asserting a claim against the Company arising pursuant to any provision of the Nevada Revised Statutes or the Company’s amended and restated articles of incorporation or Second Amended and Restated Bylaws (as either might be amended from time to time), or (d) any action or proceeding asserting a claim against the Company governed by the internal affairs doctrine. This exclusive forum provision is not applicable to any action brought under the Securities Act of 1933, as amended or The Securities Exchange Act of 1934, as amended.
We believe the choice-of-forum provision in our Second and Restated Articles of Incorporation provide for the orderly, efficient, and cost-effective resolution of Nevada-law issues affecting us by designating courts located in the State of Nevada (our state of incorporation) as the exclusive forum for cases involving such issues. However, this provision may limit a shareholder’s ability to bring a claim in a judicial forum that it believes to be favorable for disputes with us or our directors, officers, employees or agents, which may discourage such actions against us and our directors, officers, employees and agents. While there is no Nevada case law addressing the enforceability of this type of provision, Nevada courts have on prior occasion found persuasive authority in Delaware case law in the absence of Nevada statutory or case law specifically addressing an issue of corporate law. The Court of Chancery of the State of Delaware ruled in June 2013 that choice-of-forum provisions of a type similar to those included in our Second Amended and Restated Articles of Incorporation are not facially invalid under corporate law and constitute valid and enforceable contractual forum selection clauses. However, if a court were to find the choice-of-forum provision in our Second Amended and Restated Articles of Incorporation prove inapplicable to, or unenforceable in respect of, one or more of the specified types of actions or proceedings, we may incur additional costs associated with resolving such matters in other jurisdictions, which could adversely affect our business, financial condition or results of operations.
The elimination of personal liability of our directors and officers under Nevada law and the existence of indemnification rights held by our directors, officers and employees may result in substantial expenses.
Our Second Amended and Restated Articles of Incorporation and our Amended and Restated Bylaws eliminate to the furthest extent permitted under Nevada law the personal liability of our directors and officers to us, our stockholders and creditors for damages as a result of any act or failure to act in his or her capacity as a director or officer. Furthermore, our Amended and Restated Articles of Incorporation, our Amended and Restated Bylaws and individual indemnification agreements that we have entered with each of our directors and officers provide that we are obligated to indemnify, subject to certain exceptions, each of our directors or officers to the fullest extent authorized by Nevada law and, subject to certain conditions, to advance the expenses incurred by any director or officer in defending any action, suit or proceeding prior to its final disposition. Those indemnification obligations could expose us to substantial expenditures to cover the cost of settlement or damage awards against our directors or officers, which we may be unable to afford. Further, those provisions and resulting costs may discourage us or our stockholders from bringing a lawsuit against any of our current or former directors or officers for such damages, even if such actions might otherwise benefit our stockholders.
| 46 |
If, after being listing on Nasdaq, we are delisted and our shares become subject to the penny stock rules, it would become more difficult to trade our shares.
The SEC has adopted rules that regulate broker-dealer practices in connection with transactions in penny stocks. Penny stocks are generally equity securities with a price of less than $5.00, other than securities registered on certain national securities exchanges or authorized for quotation on certain automated quotation systems, provided that current price and volume information with respect to transactions in such securities is provided by the exchange or system. If we do not maintain a listing on Nasdaq and if the price of our common stock is less than $5.00, our common stock will be deemed a penny stock. The penny stock rules require a broker-dealer, before a transaction in a penny stock not otherwise exempt from those rules, to deliver a standardized risk disclosure document containing specified information. In addition, the penny stock rules require that before effecting any transaction in a penny stock not otherwise exempt from those rules, a broker-dealer must make a special written determination that the penny stock is a suitable investment for the purchaser and receive (i) the purchaser’s written acknowledgment of the receipt of a risk disclosure statement; (ii) a written agreement to transactions involving penny stocks; and (iii) a signed and dated copy of a written suitability statement. These disclosure requirements may have the effect of reducing the trading activity in the secondary market for our common stock, and therefore stockholders may have difficulty selling their shares.
If our securities are listed on Nasdaq, our failure to meet the continued listing requirements of the Nasdaq could result in a de-listing of our securities.
If after listing we fail to satisfy the continued listing requirements of Nasdaq, such as the corporate governance requirements or the minimum closing bid price requirement, Nasdaq may take steps to de-list our securities. Such a de-listing would likely have a negative effect on the price of our common stock and would impair your ability to sell or purchase our common stock when you wish to do so. In the event of a de-listing, we would take actions to restore our compliance with Nasdaq’s/ listing requirements, but we can provide no assurance that any such action taken by us would allow our common stock to become listed again, stabilize the market price or improve the liquidity of our securities, prevent our common stock from dropping below the Nasdaq minimum bid price requirement or prevent future non-compliance with Nasdaq’s/ listing requirements.
We do not intend to pay cash dividends on our capital stock in the foreseeable future.
We have never declared or paid any cash dividends on our common stock and do not anticipate paying any dividends in the foreseeable future. We currently intend to retain all future earnings to fund the development of our products.
Risks Relating to this Offering
Future sales of shares by existing stockholders could cause our share price to decline.
Sales of substantial amounts of our common stock in the public market following this offering, or the perception that these sales could occur, may cause the market price of our common stock to decline. Upon completion of this offering, we will have outstanding shares of common stock. All of the shares sold pursuant to this offering will be immediately tradable without restriction under the Securities Act, unless held by “affiliates,” as that term is defined in Rule 144 under the Securities Act. The remaining shares of common stock will be “restricted securities” within the meaning of Rule 144 under the Securities Act, but will be eligible for resale subject to applicable volume, means of sale, holding period and other limitations of Rule 144 or pursuant to an exemption from registration under the Securities Act, subject to any restrictions on unvested shares issued under our share incentive plans and subject to the terms of the lock-up agreements described below. The Underwriter may, in its sole discretion and at any time without notice, release all or any portion of the securities subject to lock-up agreements entered into in connection with this offering. See “Underwriting.”
| 47 |
Our executive officers and directors have agreed with the underwriter to a “lock-up,” meaning that, subject to certain exceptions, we, our executive officers and directors and our direct affiliates will not dispose of or hedge any of their common stock or securities convertible into or exchangeable for shares of common stock during the period from the date of this prospectus continuing through the date [_] days after the date of this prospectus, except with the prior written consent of the underwriter. Following the expiration of this [_]-day lock-up period, [ ] shares of common stock will be eligible for future sale, subject to the applicable volume, manner of sale, holding period and other limitations of Rule 144 and subject to any restrictions on unvested shares issued under our share incentive plan. Such sales may not be subject to the volume, manner of sale, holding period and other limitations of Rule 144. As resale restrictions end, the market price of our common stock could decline if the holders of those shares sell them or are perceived by the market as intending to sell them.
In the future, we may issue additional shares of common stock or other securities convertible into or exchangeable for shares of our common stock. Any of these issuances could result in substantial dilution to our existing stockholders and could cause the trading price of our common stock to decline.
Investors in this offering will experience immediate and substantial dilution of dollars per share.
You will incur immediate and substantial dilution as a result of this offering. After giving effect to the sale by us of [_] Units in this offering at a public offering price of $[_] per Unit, after deducting underwriter discounts and commissions and estimated offering expenses payable by us, investors in this offering can expect an immediate dilution of $ per Unit. For a further description of the dilution that investors in this offering may experience, see “Dilution.”
In the past, we have issued shares of common stock and warrants in private placements of our securities, and we have issued shares of common stock as compensation to our officers and directors. Our issuance of shares of common stock in the future, and the exercise of outstanding warrants or warrants that we may issue in the future, may result in additional dilution to investors in this offering.
If securities or industry analysts do not publish research or publish inaccurate or unfavorable research about our business, our stock price and trading volume could decline.
The trading market will be influenced to some extent by the research and reports that industry or financial analysts publish about us and our business. We do not control these analysts. As a small public company, we may be slow to attract research coverage and the analysts who publish information about our common stock will have had relatively little experience with us or our business and products, which could affect their ability to accurately forecast our results and could make it more likely that we fail to meet their estimates and expectations. In the event we obtain securities or industry analyst coverage, if any of the analysts who cover us provide inaccurate or unfavorable research or issue an adverse opinion regarding our stock price, our stock price could decline. If one or more of these analysts cease coverage of us or fail to publish reports covering us regularly, we could lose visibility in the market, which in turn could cause our stock price or trading volume to decline and result in the loss of all or a part of your investment in us.
Provisions in our corporate charter documents and under Nevada law could discourage another company from acquiring us and may prevent attempts by our stockholders to replace or remove our current management.
Provisions in our certificate of incorporation and our bylaws could discourage, delay or prevent a merger, acquisition or other change in control of us that stockholders may consider favorable, including transactions in which stockholders might otherwise receive a premium for their shares. These provisions could also limit the price that investors might be willing to pay in the future for shares of our common stock, thereby depressing the market price of our common stock. In addition, these provisions may frustrate or prevent any attempts by our stockholders to replace or remove our current management by making it more difficult for stockholders to replace members of our board of directors. As our board of directors is responsible for appointing the members of our management team, these provisions could in turn affect any attempt by our stockholders to replace current members of our management team. These provisions provide, among other things, that:
| ● | our board of directors is divided into three classes, Class A, Class B and Class C, with each class serving staggered three-year terms, which may delay the ability of stockholders to change the membership of a majority of our board of directors; | |
| ● | our board of directors may alter certain provisions of our amended and restated bylaws without obtaining stockholder approval; and | |
| ● | stockholders must provide advance notice and additional disclosures to nominate individuals for election to the board of directors or to propose matters that can be acted upon at a stockholders’ meeting, which may discourage or deter a potential acquirer from conducting a solicitation of proxies to elect the acquirer’s own slate of directors or otherwise attempting to obtain voting control of our shares. |
| 48 |
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
This prospectus and any documents we incorporate by reference herein may contain “forward-looking statements” (within the meaning of Section 27A of the Securities Act and Section 21E of the Securities and Exchange Act of 1934, as amended, or the Exchange Act. All statements other than statements of historical facts contained in this prospectus, including statements regarding the timing of our clinical trials, our strategy, future operations, future financial position, future revenue, projected costs, prospects, plans, objectives of management and expected market growth are forward-looking statements. These statements involve known and unknown risks, uncertainties and other important factors that may cause our actual results, performance or achievements to be materially different from any future results, performance or achievements expressed or implied by the forward-looking statements.
The Company and its representatives may from time to time make written or oral forward-looking statements with respect to the Company’s annual or long-term goals, including statements contained in its filings with the SEC and in its reports to stockholders.
The words or phrases “will likely result”, “will be”, “will”, “are expected to”, “will continue to”, “is anticipated”, “estimate”, “project” or similar expressions identify “forward-looking statements” within the meaning of the Private Securities Litigation Reform Act of 1995. Such statements are subject to certain risks and uncertainties that could cause actual results to differ materially from historical earnings and those presently anticipated or projected. Readers are cautioned not to place undue reliance on any such forward-looking statements, which speak only as of the date made.
These forward-looking statements include, among other things, statements about:
| ● | the accuracy of our estimates regarding expenses, future revenues, uses of cash, capital requirements and the need for additional financing; | |
| ● | the initiation, cost, timing, progress and results of our development activities, usability studies, preclinical studies and any clinical trials that we may be required to undertake; | |
| ● | the timing of and our ability to obtain and maintain regulatory approval of our existing product candidates, any product candidates that we may develop, and any related restrictions and/or limitations; | |
| ● | our plans to research, develop and commercialize our current and future product candidates; | |
| ● | our ability to attract collaborators with development, regulatory and commercialization expertise; | |
| ● | our ability to obtain and maintain intellectual property protection for our product candidates; | |
| ● | our ability to successfully commercialize our product candidates; | |
| ● | the size and growth of the markets for our product candidates and our ability to serve those markets; | |
| ● | the rate and degree of market acceptance of any future products; | |
| ● | the success of competing devices that are or may become available; | |
| ● | regulatory developments in the United States and other countries; | |
| ● | the performance of our third-party suppliers and manufacturers and our ability to obtain alternative sources of raw materials; | |
| ● | the impact of global inflationary pressures; |
| 49 |
| ● | our ability to obtain additional financing; | |
| ● | our use of the proceeds from our securities offerings; | |
| ● | any restrictions on our ability to use our net operating loss carry-forwards; | |
| ● | the impact of the Israel-Hamas war on our results, including potential economic restrictions imposed on and political and military instability in Israel; and | |
| ● | our ability to attract and retain key personnel. |
Moreover, we operate in a very competitive and rapidly changing environment. New risks emerge from time to time, and it is neither possible for us to predict all risk factors nor address the impact of all factors on our business or the extent to which any factor, or combination of factors, may cause our actual results to differ materially from those contained in any forward-looking statements. Actual results could differ materially from our forward-looking statements due to a number of factors, including the early stage of our product candidates presently under development; our ability to obtain and, if obtained, maintain regulatory approval of our current product candidates and any of our other future product candidates; our need for substantial additional funds in order to continue our operations and the uncertainty of whether we will be able to obtain the funding we need; our future financial performance; our ability to retain or hire key scientific or management personnel; our ability to protect our intellectual property rights that are valuable to our business, including patent and other intellectual property rights; our dependence on third-party manufacturers, suppliers, research organizations, testing laboratories and other potential collaborators; the success of our collaborations with third parties; the size and growth of the potential markets for any of our approved product candidates and the rate and degree of market acceptance of any of our approved product candidates; competition in our industry; regulatory developments in the United States and foreign countries, including the FDA; and the expected impact of new accounting standards.
You should not place undue reliance on any forward-looking statement, each of which applies only as of the date of this prospectus. Before you invest in our securities, you should be aware that the occurrence of the events described in the section entitled “Risk Factors” and elsewhere in this prospectus could negatively affect our business, operating results, financial condition and stock price. All forward-looking statements included in this document are based on information available to us on the date hereof, and except as required by law, we undertake no obligation to update or revise publicly any of the forward-looking statements after the date of this prospectus to conform our statements to actual results or changed expectations.
As used in this prospectus unless the context indicates or otherwise requires, “our Company”, “the Company”, “IR-Med”, “we”, “us” and “our” refer to IR-Med, Inc., a Nevada corporation, and its consolidated subsidiary, IR. Med Ltd., a company organized under the laws of Israel.
MARKET AND INDUSTRY DATA
This prospectus contains estimates, projections and other information concerning our industry, our business, as well as data regarding market research, estimates and forecasts prepared by our management. Information that is based on estimates, forecasts, projections, market research or similar methodologies is inherently subject to uncertainties and actual events or circumstances may differ materially from events and circumstances that are assumed in this information. The industry in which we operate is subject to a high degree of uncertainty and risk due to a variety of factors, including those described in the section titled “Risk Factors.” Unless otherwise expressly stated, we obtained this industry, business, market and other data from reports, research surveys, studies and similar data prepared by market research firms and other third parties, industry, medical and general publications, government data and similar sources. In some cases, we do not expressly refer to the sources from which this data is derived. In that regard, when we refer to one or more sources of this type of data in any paragraph, you should assume that other data of this type appearing in the same paragraph is derived from the same sources, unless otherwise expressly stated or the context otherwise requires.
| 50 |
USE OF PROCEEDS
We estimate that the net proceeds to us from this offering will be approximately $[ ] million, based on an assumed public offering price of $[ ] per Unit as set forth on the cover page of this prospectus, after deducting estimated underwriting discounts and commissions and estimated offering expenses payable by us and excluding the proceeds, if any, from the exercise of the warrants issued as part of the Units. If the underwriters’ over-allotment option is exercised in full, we estimate that our net proceeds will be approximately $[_] after deducting estimated underwriting discounts and estimated offering expenses payable by us and excluding the proceeds, if any, from the exercise of the warrants issued as part of the Units.
The principal purposes of this offering are to fund the development efforts of our products, while mainly focusing on the DiaSafe device, production of commercial units, marketing, and working capital. While there are no acquisition opportunities currently being contemplated nor have, we made an acquisition in the last three years, it is possible that an acquisition opportunity could arise that meets our strategic and financial internal return requirements. This expected use of the net proceeds from this offering represents our intentions based on our current plans and business conditions, which could change in the future as our plans and business conditions evolve. Our management will have broad discretion over the use of the net proceeds from this offering, and our investors will be relying on the judgment of our management regarding the application of the net proceeds of this offering.
Pending the use of the net proceeds from this offering as described above, we may invest the net proceeds in a variety of capital preservation instruments, including short-term, interest-bearing obligations, investment-grade instruments, certificates of deposit or direct or guaranteed obligations of the U.S. government.
A $1.00 increase (decrease) in the assumed public offering price of $[_] per Unit would increase (decrease) the amount of proceeds to us from this offering available by approximately $[_] million, assuming the number of shares offered by us, as set forth on the cover page of this prospectus, remains the same, and after deducting the estimated underwriting discounts and commissions and estimated offering expenses payable by us.
Each 1,000,000 increase (decrease) in the number of Units offered in this offering would increase (decrease) the amount of proceeds to us from this offering by approximately $[_] million, assuming that the price per share for the offering remains at $[_] and after deducting the estimated underwriting discounts and commissions and estimated offering expenses payable by us.
| 51 |
DIVIDEND POLICY
We have never declared or paid cash dividends on our capital stock, and we do not currently intend to pay any cash dividends on our capital stock in the foreseeable future. We currently intend to retain all available funds and any future earnings to fund the development and expansion of our business. Any future determination related to dividend policy will be made at the discretion of our board of directors, subject to applicable laws, and will depend upon, among other factors, our results of operations, financial condition, contractual restrictions and capital requirements. In addition, our ability to pay cash dividends on our capital stock may be limited by the terms of any future debt or preferred securities we issue or any credit facilities we enter into.
| 52 |
CAPITALIZATION
The following table sets forth our cash and restricted cash, and our capitalization as of March 31, 2024 on:
| ● | an actual basis; | |
| ● | an as adjusted basis, to reflect the sale of $[_] Units in this offering, at the assumed offering price of $[_] per Unit, and after deducting estimated underwriting discounts and commissions and estimated offering expenses payable by us. |
You should read this table together with the sections titled “Financial Data,” “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and our consolidated financial statements and the related notes included elsewhere in this prospectus. The as adjusted information below is illustrative only and our capitalization following the closing of this offering will be adjusted based on the actual public offering price and other terms of this offering determined at pricing.
As of March 31, 2024 | ||||||||
| (in thousands, except share and per share amounts) | Actual | As Adjusted | ||||||
| Cash and restricted cash | $ | 419 | ||||||
| Stockholders’ (deficiency) equity: | ||||||||
| Common stock, par value $0.001 per share, 250,000,000 shares authorized: 69,931,056 and 68,808,970 issued and outstanding as of March 31 ,2024 and December 31, 2023 | 69 | |||||||
| Additional paid-in capital | 15,341 | |||||||
| Accumulated other comprehensive loss | ) | |||||||
| Accumulated deficit | (15,496 | ) | ) | |||||
| Total stockholders’ (deficiency) equity | (86 | ) | ||||||
| Total capitalization | $ | 31,411 | ||||||
Each $1.00 increase or decrease in the assumed public offering price of $[_] per Unit would increase or decrease, as applicable, our cash and restricted cash, total stockholders’ equity and total capitalization on a pro forma basis by approximately $[_] million, assuming that the number of Units offered by us, as set forth on the cover page of this prospectus, remains the same and after deducting estimated underwriting discounts and commissions payable by us. Each increase or decrease of 1 million Units offered by us would increase or decrease our cash and restricted cash, total stockholders’ equity and total capitalization by approximately $[_] million, assuming a public offering price of $[_] per Unit and after deducting estimated underwriting discounts and commissions payable by us.
The number of shares of common stock outstanding, on an actual and pro forma basis, is based on an aggregate of 70,699,144 shares outstanding as of March 31, 2024, and excludes:
| ● | 14,096,675 shares of our common stock issuable upon the exercise of stock options as of March 31, 2024, at a weighted-average exercise price of $0.42 per share; | |
| ● | 11,330,259 shares of our common stock issuable upon the exercise of warrants as of March 31, 2024; and | |
| ● | 3,403,325 shares of our common stock that remain available for issuance as of March 31, 2024 under our 2020 Incentive Stock Plan. |
| 53 |
DILUTION
If you invest in our Units in this offering, your ownership interest will be diluted to the extent of the difference between the public offering price per share of our common stock that is part of the Unit and the pro forma as adjusted net tangible book value per share of our common stock after this offering.
Our historical negative net tangible book value as of March 31, 2024 was $86,000, or $0.001 per share of common stock based on 69,931,056 shares of common stock outstanding as of such date. Our historical net tangible book value represents our total tangible assets fewer total liabilities divided by the number of shares of our common stock outstanding as of March 31, 2024.
After giving effect to the issuance and sale of [ ] Units in this offering at an assumed public offering price of $[ ] per Unit as set forth on the cover page of this prospectus, and after deducting the estimated underwriting discounts and commissions and estimated offering expenses payable by us, our as adjusted net tangible book value as of March 31, 2024 would have been $[ ], or $[ ] per share. This represents an immediate increase/decrease in as adjusted net tangible book value of $[ ] per share to our existing stockholders and an immediate dilution of $[ ] per share to new investors purchasing shares of our common stock in this offering. We determine dilution per share to new investors by subtracting our as adjusted net tangible book value per share after this offering from the assumed public offering price per share paid by new investors in this offering.
The following table illustrates this dilution on a per share basis:
| Assumed public offering price per share | $ | |||
| Historical net tangible book value per share as of March 31, 2024 | ||||
| Decrease in historical net tangible book deficit per share attributable to pro forma transactions and other adjustments described above | ||||
| As adjusted net tangible book value per share as of [_], 2024 | ||||
| Increase in as adjusted net tangible book value per share attributable to new investors participating in this offering | ||||
| Pro forma as adjusted net tangible book value per share after this offering | ||||
| Dilution per share to new investors participating in this offering | $ |
Each $1.00 increase (decrease) in the assumed public offering price of $[_] per Unit, as set forth on the cover page of this prospectus) would increase (decrease) our pro forma as adjusted net tangible book value per share after this offering by $[_] per Unit and the dilution per share to new investors participating in this offering by $[_] per Unit, assuming that the number of Units offered by us, as set forth on the cover page of this prospectus, remains the same and after deducting estimated underwriting discounts and commissions and estimated offering expenses payable by us.
MANAGEMENT’S
DISCUSSION AND ANALYSIS OF
FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following section contains certain forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995 and other Federal securities laws and is subject to the safe-harbor created by such Act and laws. In some cases, you can identify forward-looking statements by terminology such as “may,” “will,” “should,” “expect,” “intend,” “plan,” “anticipate,” “believe,” “estimate,” “predict,” “potential” or “continue,” the negative of such terms, or other variations thereon or comparable terminology. The statements herein and their implications are merely predictions and therefore inherently subject to known and unknown risks, uncertainties, assumptions and other factors that may cause actual results, performance levels of activity, or our achievements, or industry results to be materially different from those contemplated by the forward-looking statements. Except as required by law, we undertake no obligation to release publicly the result of any revision to these forward-looking statements that may be made to reflect events or circumstances after the date hereof or to reflect the occurrence of unanticipated events. Further information on potential factors that could affect our business is described under the heading “Risk Factors” in Part I, Item 1A, of our 2023 Annual Report on Form 10-K/A for the fiscal year ended December 31, 2023 as filed with the SEC, on April 8, 2024. See also “Cautionary Note Regarding Forward-Looking Statements”.
| 54 |
Overview
We were incorporated in the State of Nevada in April 2007 under the name “Monster Motors, Inc.” We began operating the business of IR. Med Ltd., an Israeli company, through a reverse acquisition on December 24, 2020. IR. Med Ltd. (an Israeli company which was founded in 2013) continues to operate as our operating subsidiary, and we are the sole stockholder of IR. Med Ltd.
We are in the process of developing a cutting-edge infrared spectroscopy and artificial intelligence (AI) analysis technology platform, as a basis for point-of-care decision support devices. The infrared spectroscopy technology allows harmless and non-invasive gathering of bio-information from a patient’s blood and tissue. Bioinformation is then analyzed using our AI process to provide healthcare professionals with decision support in the detection and monitoring of various disease conditions.
PressureSafe, our first product based on this platform, is a handheld device designed to revolutionize early detection of pressure injuries (PIs) affecting skin and underlying tissue. PIs in the U.S. alone account for $26.8 billion in healthcare spending and result in 60,000 deaths annually. PressureSafe is expected to contribute to early detection of PIs, regardless of patient skin tone. This will drive equitable healthcare and help reduce the toll and cost of PIs. We plan to launch PressureSafe as a decision support system (DSS) tool for care givers in hospitals, nursing homes and home-care companies. On April 9, 2024, the PressureSafe decision support device received an FDA listing certification. PressureSafe is classified as a Class I device. We are currently working on completing the development of the commercial version of the PressureSafe device, planned to be launched during September 2024, following the listing under the FDA.
DiaSafe: Similarities in the physiological development of PIs and diabetic foot ulcers (“DFU”) under the skin surface allow the IRMED PressureSafe device to be adopted to support the early detection of DFU among diabetic patients at high risk of developing DFU. We are assessing and planning the development of our second product, which is a handheld optical monitoring device that will support early detection of DFUs in lower limb skin and underlying tissue, primarily caused by prolonged pressure on the sole among diabetic patients a condition, which sometimes is accompanied by other comorbidities as lower limb neuropathy.
Our novel technology platform will enable direct assessment of the development of a DFU before it becomes an open wound that may lead to limb amputation. The Israeli Innovation Authority, or IIA, has approved our plan to develop a diabetic foot ulcer device for early detection of DFU. On January 25, 2024, the IIA approved a program to develop a device for the early detection of diabetic foot ulcers among diabetic patients, with a project budget of NIS 3,761,978 (approximately US$ 1,030,000) which includes an amount equal to 50% grant of the total budget provided at the time of the grant, disbursed in installments over the course of 13 months, by the project’s progress. In consideration for the grant by the IIA, the subsidiary is required to pay royalties at the rate of 3%-5% from the total sales until the repayment date of the full amount of the grant, plus annual interest at the SOFR rate. In addition, the IIA must approve any arrangement whereby the Company seeks to transfer the technology relating to the project, or its development, from Israel. Following the IIA grant we plan to commence a clinical trial in the center of Israel’s leading diabetes clinic.
Our initial focus is on the development of decision support system solutions utilizing our proprietary platform for the pre-emptive diagnosis of PIs, diabetic foot ulcers, and mid-ear infections detection. Our current business plan focuses on two principal medical devices currently in development:
| 1. | PressureSafe, a handheld optical monitoring device that is being developed to support early detection of PIs to the skin and underlying tissue, primarily caused by prolonged pressure associated with bed confinement; and | |
| 2. | DiaSafe, a handheld optical monitoring device that is being developed to support early detection of DFUs in lower limb skin and underlying tissue, primarily caused by prolonged pressure on the sole of the foot and diabetes. |
| 55 |
Our product candidates are in various stages of development. The PressureSafe device is in an advanced stage of development and is planned to be our first go-to-market product, while the DiaSafe device is in the initial stages of research and development.
We have completed the development of the first generation PressureSafe prototype in the second quarter of 2022. In June 2022, IR. Med Ltd., our wholly owned subsidiary, entered into a study agreement with Beit Rivka, a large geriatric hospital in Israel associated with Clalit, the largest Health Maintenance Organization (HMO) in Israel and second largest globally, to conduct a usability study of PressureSafe.
On July 17, 2023, we published our interim report of usability study performed in Israel in leading medical centers with the following results: PressureSafe demonstrated very high efficacy in noninvasively detecting the presence and absence of PIs below the skin’s surface. PressureSafe accurately detected the presence of a PI in 96% of cases. In addition, PressureSafe correctly determined that no wound was present in 91% of cases. The study was conducted at two medical centers owned by Clalit, namely Beit Rivka Hospital and Rabin Medical Center, both in Petah Tikva, where 370 PressureSafe scans were performed on 25 patients who had Stage 1 PIs or deep tissue injuries. No device related safety issues were reported in the total of 44 patients evaluated for safety.
On September 26, 2023, we announced that we signed a Clinical Trial Agreement with the Methodist Healthcare System of San Antonio to conduct a useability study titled “Safety and Efficacy of the PressureSafe Device for Early Detection of Pressure Injury in People with Various Skin Tones, Including Dark Skin Tones.” Methodist Healthcare is recognized as the most respected healthcare provider in its region. With a network of 85 hospitals, 9 of which are acute care facilities, Methodist Healthcare employs more than 11,000 people, including 2,700 physicians. Approximately 50% of subjects recruited for the upcoming study will have dark skin tone, thus producing comparative data on PressureSafe’s accuracy as a decision support device in detecting early-stage PIs in people of darker and lighter skin tones. Early-stage PIs can be more difficult to see on dark skin tones using the current standard of care for the detection of PIs, which is visual skin inspection.
On February 20, 2024, we announced highly favorable proof of efficacy data for PressureSafe. Data from the study conducted at two medical centers owned by Clalit, Beit Rivka Hospital and Rabin Medical Center, presented at the National Pressure Injury Advisory Panel (NPIAP) 2024 Annual Conference on February 16 and 17, 2024 in San Antonio, Texas. Dr. Gal Maydan of Beit Rivka Hospital Geriatric Rehabilitation Center, and Principal Investigator of the study, presented the data in a poster titled “Near Infra-Red Spectroscopy for early detection of stage 1 pressure injury and deep tissue injury – clinical study results”. While the current standard of care for the detection of pressure injuries is visual and tactile clinical evaluation, physiological changes below the skin’s surface, including inflammation and interstitial fluids precede changes on the surface. The objective of the study was to evaluate the sensitivity, specificity, and usability of PressureSafe to detect early-stage pressure injuries Stage 1 suspected deep tissue injuries (sDTI) before skin breakage, compared to standard of care. PressureSafe detected biomarkers and changes in tissue structures under the skin’s surface as they relate to pressure injuries.
The 14-day efficacy portion of the single arm, bi-center study evaluated 38 patients at high risk of pressure injury development. A total of 924 scans were conducted on 154 body locations. Nurses conducting the scans were blinded to PressureSafe’s results, which were encrypted. PressureSafe detected Stage 1 pressure injuries with 92% sensitivity and 88% specificity. Additional portions of the study evaluated safety, as well as device calibration and validation. Total data from 66 patients was obtained for safety analysis and no safety signals were identified in 1,493 scans. Based on these data, the study concluded that PressureSafe is a safe, efficient, and valuable method for early detection of pressure injuries.
On April 9, 2024, the PressureSafe decision support device received an FDA listing certification. PressureSafe is classified as a Class I device. We are currently working on completing the development of the commercial version of the PressureSafe device, planned to be launched during September 2024, following the listing under the FDA.
Key Financial Terms and Metrics
The following discussion summarizes the key factors our management believes are necessary for an understanding of our consolidated financial statements.
| 56 |
Revenues
We have not generated any revenues from product sales to date.
Research and Development Expenses
The process of researching and developing our product candidates is lengthy, unpredictable and subject to many risks. We expect to continue incurring substantial expenses for the next several years as we continue to develop our product candidates. We are unable, with any certainty, to estimate either the costs or the timelines in which those expenses will be incurred. Our current development plans focus on the development of our PressureSafe and DiaSafe diagnostic devices. The design and development of these devices will consume a large proportion of our current, as well as projected, resources.
Our research and development costs are comprised of:
| ● | internal recurring costs, such as personnel-related costs (salaries, employee benefits, equity compensation and other costs), materials and supplies, facilities and maintenance costs attributable to research and development functions; and | |
| ● | fees paid to external parties who provide us with contract services, such as preclinical testing, manufacturing, related testing and clinical trial activities. |
Marketing
Marketing expenses consist primarily of salaries, employee benefits, equity compensation and other personnel-related costs associated with executive and other support staff. Other significant marketing expenses include the costs associated with professional fees to develop our marketing strategy.
General and Administrative Expenses
General and administrative expenses consist primarily of salaries, employee benefits, equity compensation and other personnel-related costs associated with executive, administrative and other support staff. Other significant general and administrative expenses include the costs associated with professional fees for accounting, auditing, insurance costs, consulting and legal services, along with facility and maintenance costs attributable to general and administrative functions.
Financial Expenses
Financial expenses consist primarily impact of exchange rate derived from re-measurement of monetary balance sheet items denominated in non-dollar currencies. Other financial expenses include bank’s fees and interest on stockholders’ loans.
| 57 |
Comparison of the Year Ended December 31, 2023, to the Year Ended December 31, 2022.
Our financial results for the year ended December 31, 2023, are summarized as follows in comparison to the year ended December 31, 2022:
| Year Ended | ||||||||
| December 31, 2023 | December 31, 2022 | |||||||
| U.S. dollars (in thousands) | ||||||||
| Operating Expenses: | ||||||||
| Research and Development | 2,061,000 | 1,885,000 | ||||||
| Marketing | 822,000 | 759,000 | ||||||
| General and Administrative | 2,028,000 | 2,118,000 | ||||||
| Financing income, net | (2,000 | ) | (28,000 | ) | ||||
| Loss for the year | 4,909,000 | 4,734,000 | ||||||
Revenues. We have not recorded any revenues to date.
Research and Development Expenses. Research and development expenses increased from $1,885,000 for the year ended December 31, 2022, to $2,061,000 for the year ended December 31, 2023. The increase resulted primarily from increased use of third-party contractors for further research and development activities, and non-cash expenses recorded relating to stock-based compensation to employees.
Marketing Expenses. Marketing expenses increased from $759,000 for the year ended December 31, 2022, to $822,000 in the year ended December 31, 2023. The increase resulted primarily from non-cash expenses attributable to stock-based compensation to service providers. This increase was partially offset by a reduction in payroll expenses and reduction in professional services.
General and Administrative Expenses. General and administrative expenses decreased from $2,118,000 for the year ended December 31, 2022, to $2,028,000 in the year ended December 31, 2023. The decrease resulted primarily from a reduction in payroll expenses and a reduction in professional services. This decrease was partially offset by the increase in non-cash expenses attributable to stock-based compensation to our directors, officers and service providers.
Loss. Loss for the year ended December 31, 2023, was $4,909,000 compared to $4,734,000 for the year ended December 31, 2022, and is primarily attributable to the increase in non-cash expenses due to stock-based compensation to directors, officers and service providers, and utilization of third-party contractors for further research and development activities. These increases were partially offset by the reduction in professional services and payroll expenses related to marketing and general and administrative activities.
Comparison of the Three Months Ended March 31, 2024, to the Three Months Ended March 31, 2023
For the three months ended March 31, | ||||||||
| 2024 | 2023 | |||||||
| U.S dollars (in thousands) | ||||||||
| Research and development expenses, net | 195 | 605 | ||||||
| Marketing expenses | 168 | 172 | ||||||
| General and administrative expenses | 295 | 575 | ||||||
| Total operating expenses | 658 | 1,352 | ||||||
| Financial income, net | (1 | ) | (2 | ) | ||||
| Loss for the period | 657 | 1,350 | ||||||
Revenues. During the three-month period ended March 31, 2024, and 2023, we did not record any revenues from operations.
| 58 |
Research and Development Expenses. Research and development expenses consist of salaries and related expenses, consulting fees, service providers’ costs, and overhead expenses. Research and development expenses decreased from $605,000 during the three months ended March 31, 2023, to $195,000 during the corresponding three-month period in 2024. The decrease in the 2024 period resulted primarily from a decrease in the use of third-party contractors for further research and development activities due to the completion of the development of the PressureSafe device, proceeds of a grant from the IIA, and non-cash expenses recorded relating to stock-based compensation to employees.
Marketing Expenses. Marketing expenses consist primarily of salaries and professional services. Marketing expenses decreased from $172,000 during the three months ended March 31, 2023, to $168,000 during the corresponding three-month period in 2024. The decrease in marketing expenses resulted primarily from the reduction in professional services, partially offset by an increase in non-cash expenses attributable to stock-based compensation to service providers.
General and Administrative Expenses. General and administrative expenses consist primarily of salaries and related expenses and other non-personnel related expenses such as legal and accounting-related expenses. General and administrative expenses decreased from $575,000 during the three months ended March 31, 2023, to $295,000 in the corresponding three-month period in 2024. The decrease in general and administrative expenses resulted primarily from a decrease in non-cash expenses attributable to stock-based compensation to our directors, officers and service providers, a reduction in payroll expenses and a reduction in professional services.
Loss. Loss for the three months ended March 31, 2023, was $1,350,000 compared to $657,000 for the corresponding three-month period in 2024. The decrease in net loss is primarily attributable to a decrease in use of third-party contractors for further research and development activities due to the completion of the development of the PressureSafe device, proceeds of a grant from the IIA, a decrease in non-cash expenses attributable to stock-based compensation to our directors, officers and service providers and a reduction in payroll expenses and professional services.
Liquidity and Capital Resources
We are subject to risks common to companies in the medical device industry, including but not limited to, the need for additional capital, the need to obtain marketing approval and reimbursement for any product candidate that we may identify and develop, the need to successfully commercialize and gain market acceptance of our product candidates, dependence on key personnel, protection of proprietary technology, compliance with government regulations, development of technological innovations by competitors, reliance on third-party manufacturers and the ability to transition from pilot-scale production to large-scale manufacturing of products.
From inception, we have funded our operations from a combination of loans and sales of equity instruments. In 2021 and 2022, we raised aggregate gross proceeds of $5,830,000 and $3,625,000, respectively, from sales of our equity and equity linked securities. In addition, on June 12, 2023, we raised aggregate gross proceeds of $1,000,000 from sales of our shares of common stock and warrants to purchase shares of common stock. On June 4, 2024, we raised aggregate gross proceeds of $715,000 from sales of our shares of common stock and warrants to purchase shares of common stock.
As of December 31, 2023, we had a total of $767,000 in cash resources and approximately $634,000 of liabilities, consisting of $473,000 of current liabilities from operations.
| 59 |
The following table provides a summary of operating, investing, and financing cash flows for the years ended December 31, 2023, and 2022 respectively (in thousands):
| Year ended | ||||||||
| December 31, 2023 | December 31, 2022 | |||||||
| US Dollars (In thousands) | ||||||||
| Net cash used in operating activities | (3,232 | ) | (3,375 | ) | ||||
| Net cash used in investment activities | (2 | ) | (59 | ) | ||||
| Net cash provided by financing activities | 1,000 | 3,625 | ||||||
As of March 31, 2024, we had $408,000 in cash resources and approximately $665,000 of liabilities, including $505,000 of current liabilities from operations.
The following table provides a summary of operating, investing, and financing cash flows for the three months ended March 31, 2024 (in thousands):
| For the three months ended | ||||||||
| March 31, 2024 | March 31, 2023 | |||||||
| US Dollars (In thousands) | ||||||||
| Net cash used in operating activities | (358 | ) | (900 | ) | ||||
We have experienced operating losses since the Company’s inception and had a total accumulated deficit of $14,839,000 as of December 31, 2023, and a total accumulated deficit of $15,484,000 as of March 31, 2024. We expect to incur additional costs and require additional capital. We have incurred losses in nearly every year since inception, for the year ended December 31, 2023, and for the three months period ended March 31, 2024. These losses have resulted in significant cash used in operations. During the years ended December 31, 2023, and 2022, our cash used in operations was approximately $3,232,000, and $3,375,000, respectively. During the three months ended March 31, 2024, and 2023, our cash used in operations was approximately $358,000 and $900,000, respectively. We need to continue and intensify our research and development efforts for our product candidates (which are in various stages of development), strengthen our patent portfolio, establish operations processes and pursue FDA clearance and international regulatory approvals. As we continue to conduct these activities, we expect the cash needed to fund operations to increase significantly over the next several years.
Under the private placement of our securities that we undertook between December 2020 and April 2021, we entered into a securities purchase agreement with certain accredited investors providing for the issuance and sale to such investors of an aggregate of 18,221,876 shares of our Common Stock and warrants for an additional 9,110,938 shares of our Common Stock, exercisable through December 24, 2023, at a per share exercise price of $0.64. After deducting for offering related expenses, the aggregate net proceeds from the initial closing of the 2020 Private Placement were approximately $5,446,000.
Under the private placement of our securities which we commenced in April 2022 through July 2022, we entered into a securities purchase agreement with six accredited investors providing for the issuance and sale to such investors of an aggregate of 4,119,321 shares of our common stock and warrants for an additional 4,119,321 shares of our common stock, exercisable through 2024, at a per share exercise price of $1.10. The Company is entitled to expedite the warrant exercise period for all or a part of the then outstanding warrants by written notice to the holders if the publicly traded price of our common stock equals or exceeds $2.50 per share (which amount may be adjusted for certain capital events, such as stock splits, as described herein) and the corresponding average daily trading volume during such period equals or exceed 75,000 shares, in each case for the forty (40) consecutive trading days. The aggregate gross proceeds from the private placement were approximately $3,625,000.
In addition, on June 12, 2023, we raised aggregate gross proceeds of $1,000,000 from sales of our shares of common stock and warrants to purchase shares of common stock.
On June 4, 2024, we raised aggregate gross proceeds of $715,000 from sales of our shares of common stock and warrants to purchase shares of common stock.
As of March 31, 2024, we had $408,000 in cash resources and approximately $665,000 of liabilities, including $505,000 of current liabilities from operations. On June 4, 2024, following the 2024 Private Placement the Company received aggregate gross proceeds of $715,000, which enables us to fund our operations requirements for the next four months.
| 60 |
We expect that the proceeds from this offering will enable us to fund our operations and capital expenditure requirements for the next 18 months.
We will need to obtain additional funding in order to pursue our business plans. If we are unable to raise capital when needed or on attractive terms, we would be forced to delay, reduce or eliminate our research and development programs or future commercialization efforts.
Our requirements for additional capital during this period will depend on many factors, including the following:
| ● | the scope, rate of progress, results and cost of our development and engineering efforts to develop the PressureSafe and DiaSafe devices, clinical studies (to the extent necessary), preliminary testing activities and other related activities; | |
| ● | the cost, timing and outcomes of regulatory related efforts for commercial sales approvals; | |
| ● | the cost and timing of establishing sales, marketing and distribution capabilities; | |
| ● | the terms and timing of any collaborative, licensing and other arrangements that we may establish; | |
| ● | the timing, receipt and amount of sales, profit sharing or royalties, if any, from our potential products; | |
| ● | the cost of preparing, filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights; and | |
| ● | the extent to which we acquire or invest in businesses, products or technologies, although we currently have no commitments or agreements relating to any of these types of transactions. |
For the year ended December 31, 2023, for the three months ended March 31, 2024, and as of the date of this report, we assessed our financial condition and concluded that based on our current and projected cash resources and commitments, as well as other factors mentioned above, there is a substantial doubt about our ability to continue as a going concern. We are planning to raise additional capital to continue our operations, as well as to explore additional avenues to increase revenues and reduce expenditures. We cannot be sure that future funding will be available to us on acceptable terms, or at all. Due to often volatile nature of the financial markets, equity and debt financing may be difficult to obtain.
We may seek to raise any necessary additional capital through a combination of private or public equity offerings, debt financings, collaborations, strategic alliances, licensing arrangements and other marketing and distribution arrangements. To the extent that we raise additional capital through marketing and distribution arrangements or other collaborations, strategic alliances or licensing arrangements with third parties, we may have to relinquish valuable rights, future revenue streams, or product candidates or to grant licenses on terms that may not be favorable to us. If we raise additional capital through private or public equity offerings, the ownership interest of our existing stockholders will be diluted, and the terms of these securities may include liquidation or other preferences that adversely affect our stockholders’ rights. If we raise additional capital through debt financing, we may be subject to covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends.
Off-Balance Sheet Arrangements
We do not have any off-balance sheet arrangements.
Critical Accounting Policies and Estimates
The preparation of financial statements in conformity with GAAP requires management to make judgments, estimates and assumptions that affect the reported amounts of assets and liabilities and disclosures of contingent assets and liabilities at the date of the consolidated financial statements, and the reported amounts of expenses during the reporting period. Actual results could differ from those estimates.
Accounting for share-based compensation
We recognize all employee and nonemployee stock-based compensation as a cost in the consolidated financial statements. For awards with a graded vesting schedule, we use the graded vesting attribution approach to recognize compensation cost over the vesting period.
| 61 |
We estimated grant date fair value using the Black-Scholes-Merton option-pricing model and estimates the number of forfeitures expected to occur.
Smaller Reporting Company Status
Currently, we qualify as a smaller reporting company.
As a smaller reporting company, we are eligible and have taken advantage of certain exemptions from various reporting requirements that are not available to public reporting companies that do not qualify for this classification, including, but not limited to:
● An opportunity for reduced disclosure obligations regarding executive compensation in our periodic and annual reports, including without limitation exemption from the requirement to provide a compensation discussion and analysis describing compensation practices and procedures,
● An opportunity for reduced financial statement disclosure in registration statements and in annual reports on Form 10-K, which only requires two years of audited financial statements rather than the three years of audited financial statements that are required for other public companies,
● An opportunity for reduced audit and other compliance expenses as we are not subject to the requirement to obtain an auditor’s report on internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act of 2002, and
● An opportunity to utilize the non-accelerated filer time-line requirements beginning with our annual report for the year ending December 31, 2023, and quarterly filings thereafter.
For as long as we continue to be a smaller reporting company, we expect that we will take advantage of both the reduced internal control audit requirements and the disclosure obligations available to us as a result of this classification.
JOBS Act Transition Period
Section 107 of the JOBS Act provides that an “emerging growth company” can take advantage of the extended transition period provided in Section 7(a)(2)(B) of the Securities Act for complying with new or revised accounting standards. Thus, an emerging growth company can delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. We have elected to take advantage of the extended transition period to comply with new or revised accounting standards and to adopt certain of the reduced disclosure requirements available to emerging growth companies. As a result of the accounting standards election, we will not be subject to the same implementation timing for new or revised accounting standards as other public companies that are not emerging growth companies which may make comparison of our financials to those of other public companies more difficult.
| 62 |
BUSINESS
Our Mission
We are developing a cutting-edge infrared spectroscopy and AI analysis technology platform as a basis for point-of-care decision support devices. The infrared spectroscopy technology allows harmless and non-invasive gathering of bio-information from patient blood and tissue. Bioinformation is then analyzed using the company’s AI process to provide healthcare professionals with decision support in the detection and monitoring of various disease conditions.
Overview
On February 28, 2024, following financial difficulties our Board of Directors resolved that the Company’s operations will be limited only to critical actions in order to save funds. Accordingly, the following description of our two product candidates’ development and commercialization plans are currently limited and are subject to us being able to raise additional funds to support our operations and to further develop and commercialize our products, which are in various stages of design and development.
We are in the process of developing a cutting-edge infrared spectroscopy and artificial intelligence (AI) analysis technology platform, as a basis for point-of-care decision support devices. The infrared spectroscopy technology allows harmless and non-invasive gathering of bio-information from a patient’s blood and tissue. Bioinformation is then analyzed using our AI process to provide healthcare professionals with decision support in the detection and monitoring of various disease conditions.
PressureSafe, our first product based on this platform, is a handheld device designed to revolutionize early detection of pressure injuries (PIs) affecting skin and underlying tissue. PIs in the U.S. alone account for $26.8 billion in healthcare spending and result in 60,000 deaths annually. PressureSafe is expected to contribute to early detection of PIs, regardless of patient skin tone. This will drive equitable healthcare and help reduce the toll and cost of PIs. We plan to launch PressureSafe as a decision support system (DSS) tool for care givers in hospitals, nursing homes and home-care companies. On April 9, 2024, the PressureSafe decision support device received an FDA listing certification. PressureSafe is classified as a Class I device. We are currently working on completing the development of the commercial version of the PressureSafe device, planned to be launched during September 2024, following the listing under the FDA.
DiaSafe: Similarities in the physiological development of PIs and DFU under the skin surface allow the IRMED PressureSafe device to be adopted to support the early detection of DFU among diabetic patients at high risk of developing DFU. We are assessing and planning the development of our second product, which is a handheld optical monitoring device that will support early detection of DFUs in lower limb skin and underlying tissue, primarily caused by prolonged pressure on the sole among diabetic patients a condition, which sometimes is accompanied by other comorbidities as lower limb neuropathy.
Based on data published by the Diabetes Research Institute:
| ● | Diabetes is increasing at an alarming rate in the U.S. According to the Centers for Disease Control’s (CDC) National Diabetes Statistics Report for 2022, cases of diabetes have risen to an estimated 37.3 million people, or 11.3% of the U.S. population. An estimated 28.7 million people have diagnosed diabetes, while approximately 8.6 million people have diabetes but have not yet been diagnosed. | |
| ● | In 2019, 283,000 children and adolescents younger than 20 years old were diagnosed with diabetes. This includes 244,000 with type 1 diabetes. |
Incidence of Diabetes Complications
| ● | Diabetes can affect many parts of the body and is associated with serious complications, such as heart disease, stroke, blindness, kidney failure and lower-limb amputation, among other conditions. | |
| ● | Risks associated with diabetes in the U.S. vary. Studies suggest that the lifetime risk of developing a diabetic foot ulcer, or DFU, is between 19% and 34% of diabetic patients. Infection develops in 50%-60% of ulcers and is a principal cause of damage in diabetic feet. Approximately 20% of moderate or severe diabetic foot infections result in lower extremity amputations. |
| 63 |
Early detection of DFU is crucial. Signs of redness (which is especially difficult to detect among darker-skin patients), blisters and minor injuries are very important to detect. Our technology platform for early detection of PIs is being adjusted to detect incipient DFUs. Applying our unique AI technology with an appropriate optical design allows different foot measurements and is expected to provide patients and caregivers an alert of potentially developing DFU in its early stages.
Our novel technology platform will enable direct assessment of the development of a DFU before it becomes an open wound that may lead to a limb amputation. The Israeli Innovation Authority, or IIA, has approved our plan to develop a diabetic foot ulcer device for early detection of DFU .On January 25, 2024, the IIA approved a program to develop a device for the early detection of diabetic foot ulcers among diabetic patients, with a project budget of NIS 3,761,978 (approximately US$ 1,030,000) which includes an amount equal to 50% grant of the total budget provided at the time of the grant, disbursed in installments over the course of 13 months, in accordance with the project’s progress. In consideration for the grant by the IIA, the subsidiary is required to pay royalties at the rate of 3%-5% from the total sales until the repayment date of the full amount of the grant, plus annual interest at the SOFR rate. In addition, the IIA must approve any arrangement whereby the Company seeks to transfer the technology relating to the project, or its development, from Israel. Following the IIA grant, we plan to commence a clinical trial in the center of Israel’s leading diabetes clinic.
Future research and development can be an innovative otoscope, Nobiotics, to support physicians with an immediate indication as to whether mid-ear infection (otitis media), a common malady in children, is of a bacterial origin and thus requiring antibiotic treatment, or of a viral origin that consequently does not require antibiotic treatment.
Our technology platform utilizes AI. AI is a broad term generally used to describe conditions where a machine mimics “cognitive” functions associated with human intelligence, such as “learning” and “problem solving.” Basic AI includes machine learning, where a machine uses algorithms to parse data, learn from it, and then suggest a determination or prediction about a given phenomenon. The machine is “trained” using large amounts of data and algorithms that provide it with the ability to learn how to perform various tasks.
The global diagnostics market is driven in large by solutions that can be applied in healthcare settings, as these tools will drive decisions regarding specific treatments and the associated outlays. However, despite advances in medical imaging and other diagnostic tools, misdiagnosis remains a common occurrence.
Our initial focus is on the development of decision support system solutions utilizing our proprietary platform for the pre-emptive diagnosis of PIs, diabetic foot ulcers. Our current business plan focuses on two principal medical devices
| 1. | PressureSafe, a handheld optical monitoring device that is being developed to support early detection of PIs to the skin and underlying tissue, primarily caused by prolonged pressure associated with bed confinement; and | |
| 2. | DiaSafe, a handheld optical monitoring device that is being developed to support early detection of DFUs in lower limb skin and underlying tissue, primarily caused by prolonged pressure on the sole of the foot and diabetes. |
Our product candidates are in various stages of development. The PressureSafe device is planned to be our first go-to-market product, while the DiaSafe device is in research and development.
We have completed the development of the first generation PressureSafe prototype in the second quarter of 2022. In June 2022, IR. Med Ltd., our wholly owned subsidiary, entered into a study agreement with Beit Rivka, a large geriatric hospital in Israel associated with Clalit, the largest Health Maintenance Organization (HMO) in Israel and second largest globally, to conduct a usability study of PressureSafe.
| 64 |
On July 17, 2023, we published our interim report of usability study performed in Israel in leading medical centers with the following results: PressureSafe demonstrated very high efficacy in noninvasively detecting the presence and absence of PIs below the skin’s surface. PressureSafe accurately detected the presence of a PI in 96% of cases. In addition, PressureSafe correctly determined that no wound was present in 91% of cases. The study was conducted at two medical centers owned by Clalit, namely Beit Rivka Hospital and Rabin Medical Center, both in Petah Tikva, where 370 PressureSafe scans were performed on 25 patients who had Stage 1 PIs or deep tissue injuries. No device related safety issues were reported in the total of 44 patients evaluated for safety.
On September 26, 2023, we announced that we signed a Clinical Trial Agreement with the Methodist Healthcare System of San Antonio to conduct a useability study titled “Safety and Efficacy of the PressureSafe Device for Early Detection of Pressure Injury in People with Various Skin Tones, Including Dark Skin Tones.” Methodist Healthcare is recognized as the most respected healthcare provider in its region. With a network of 85 hospitals, 9 of which are acute care facilities, Methodist Healthcare employs more than 11,000 people, including 2,700 physicians. Approximately 50% of subjects recruited for the upcoming study will have dark skin tone, thus producing comparative data on PressureSafe’s accuracy as a decision support device in detecting early-stage PIs in people of darker and lighter skin tones. Early-stage PIs can be more difficult to see on dark skin tones using the current standard of care for the detection of PIs, which is visual skin inspection.
On February 20, 2024, we reported 92% efficacy for PressureSafe. Data from the study conducted at two medical centers owned by Clalit, the world’s second largest health maintenance organization (HMO) and the largest in Israel, Beit Rivka Hospital and Rabin Medical Center, presented at the National Pressure Injury Advisory Panel (NPIAP) 2024 Annual Conference on February 16 and 17, 2024 in San Antonio, Texas. The 14-day efficacy portion of the single arm, bi-center study evaluated 38 patients at high risk of pressure injury development. A total of 924 scans were conducted on 154 body locations. Nurses conducting the scans were blinded to PressureSafe’s results, which were encrypted. PressureSafe detected Stage 1 pressure injuries with 92% sensitivity and 88% specificity. Additional portions of the study evaluated safety, as well as device calibration and validation. Total data from 66 patients was obtained for safety analysis and no safety signals were identified in 1,493 scans. Based on these data, the study concluded that PressureSafe is a safe, efficient, and valuable method for early detection of pressure injuries.
On April 9, 2024, the PressureSafe decision support device received an FDA listing certification. PressureSafe is classified as a Class I device. We are currently working on completing the development of the commercial version of the PressureSafe device, planned to be launched during September 2024, following the listing under the FDA.
Overview of Target Market and our Solutions
Pressure Injury Market
Populations are aging due to improvements in healthcare. However, there are increased rates of obesity, diabetes and cardiovascular diseases. This combination of an increasingly aging population and such diseases has resulted in more people with decreased mobility needing assistance with activities of daily living. A major morbidity of decreased mobility is development of PIs. PIs develop as a result of a combination of physiologic events and external conditions. Along with localized ischemia and reperfusion injury to tissues, impaired lymphatic drainage and mechanical deformation of tissue cells have been shown to contribute to injury.
Compression prevents lymph fluid drainage and leads to deterioration in tissue cell normal activities, which causes increased interstitial fluid and waste build up, contributing to the development of PIs. The time required to develop PIs depends on many factors, including the patient’s physiology and the degree of pressure and sheer force placed on the tissue. PIs occur over predictable pressure points where bony protuberances are more likely to compress tissues when the patient is in prolonged contact with hard surfaces. Studies show that the heel area is the second most frequent location for a pressure ulcer, with the most prevalent being the sacrum. The heel accounts for between 23% and 28% of all pressure ulcers.12
12 Smith, S., Ashby, S., Thomas, L. and Williams, F., 2017. Evaluation of a multifactorial approach to reduce the prevalence of pressure injuries in regional Australian acute inpatient care settings. International Wound Journal, 15(1), pp.95-105.
| 65 |
While the overall number of Hospital Acquired Conditions (HAC) has decreased by 8%,13 pressure injuries have resisted improvement efforts and continue to grow by 10% annually. PIs are both costly and deadly. The U.S. Agency for Healthcare Research and Quality (AHRQ) reports that PIs add $10.2 billion to annual U.S. healthcare costs.14 Furthermore, these are associated with over 45% of the 63,619 HAC related deaths in the U.S., making it the leading HAC related death.
PIs impose a tremendous healthcare burden. As stated in the National Pressure Injury Advisory Panel fact sheet for 2023, 60,000 patients die every year as a direct result of pressure injuries. Acute care attributable to hospital-acquired PIs reaches $26.8 billion, and 2.5 million patients per year develop a PI. Patient care costs per PI range from $20,900 up to $151,700. PIs are among one of the five most common harms experienced by patients and the second most common claim for lawsuits, after wrongful death. PIs occur across the healthcare industry, including in 10% of acute care patients, 25% of long-term acute care patients, 12% of nursing home patients and 12% of rehabilitation center patients.
The most common method used to detect early PIs is a visual assessment by a professional caregiver focusing on areas where PIs most frequently develop. This visual assessment is subjective, unreliable, untimely (as PIs often occur suddenly without visual cues), ineffective and only effective to detect PIs once they are visible. Technology-based methods for detecting and monitoring have been developed, but as far as we know, none have succeeded in providing an effective solution.
Pressure Injuries Background
Currently, PIs are discovered only as they begin to appear on the skin, after they have been festering underneath the skin layers. Nurses regularly assess patients at high risk by evaluating them according to accepted scores (e.g., Braden or Norton Scales). Hospitals can then get the patient onto a different type of mattress that wicks away moisture and reduces pressure and impose orders for the individual to be turned every 2 hours, for example. The risk of a PI among ICU patients ranges between 14%-17% of patients.15
Intrinsic risk factors such as diabetes, malnutrition and smoking also increase the overall risk for pressure injuries. The spinal cord injury patient population is at the highest risk (25-66%) of developing a PI due to the combination of immobility and decreased sensation. A prospective study of spinal cord patients not only found that sacral and ischial PIs were very common (43% and 15%, respectively), as might be expected, but also noted that the second most common location was on the heel (19%).16
Nursing home patients have PI prevalence of 11%17 and are most likely to develop PIs on the sacrum or heels. Nursing home patients were also found to have contractures at a prevalence of 55%. Contractures are caused by decreased elasticity of the tissue surrounding major joints, and the resulting lack of full mobility in the affected extremities significantly increases the risk of PI information.
A significant market is the home healthcare market, which is anticipated to be worth $645 billion by 2025 (CAGR 8.7%).18 It is estimated that by 2030, seniors aged 65 and over will represent 20% of the U.S. population, and over 19 million seniors are estimated to need homecare services. Homecare companies have a strong incentive to prevent PIs as they are rated and carry part of the cost treating those patients.
13 AP News. 2019. Pressure Ulcers Cost U.S. Healthcare $10.2 Billion and Contribute to Nearly 29,000 Hospital Deaths Each Year.
14 Boyko, T., Longaker, M. and Yang, G., 2018. Review of the Current Management of Pressure Ulcers. Advances in Wound Care, 7(2), pp.57-67.
15 Fowler, E., Scott-Williams, S. and McGuire, J., 2008. Practice Recommendations for Preventing Heel Pressure Ulcers. Ostomy Wound Management, 54(10), pp.42-57.
16 Delmore, B., Lebovits, S., Suggs, B., Rolnitzky, L. and Ayello, E., 2015. Risk Factors Associated with Heel Pressure Ulcers in Hospitalized Patients. Journal of Wound, Ostomy & Continence Nursing, 42(3), pp.242-248; Vecin NM, Gater DR. Pressure Injuries and Management after Spinal Cord Injury. J Pers Med. 2022 Jul 12;12(7):1130. doi: 10.3390/jpm12071130. PMID: 35887627; PMCID: PMC9325194; Boyko TV, Longaker MT, Yang GP. Review of the Current Management of Pressure Ulcers. Adv Wound Care (New Rochelle). 2018 Feb 1;7(2):57-67. doi: 10.1089/wound.2016.0697. PMID: 29392094; PMCID: PMC5792240.
17 Palese, A., Zammattio, E., Zuttion, R., Ferrario, B., Ponta, S., Gonella, S. and Comoretto, R., 2020. Avoidable and Unavoidable Pressure Injuries Among Residents Living in Nursing Homes. Journal of Wound, Ostomy & Continence Nursing, 47(3), pp.230-235.
18 Ferrell, B., Josephson, K., Norvid, P. and Alcorn, H., 2015. Pressure Ulcers Among Patients Admitted to Home Care. Journal of the American Geriatrics Society, 48(9), pp.1042-1047.
| 66 |
According to a survey published in 2000 by UCLA School of Medicine,19 in a total sample of 3,048 patients, 9.12% had PIs, and of these, 37.4% had more than one PI, and 14% had three or more. Considering the worst PIs for each subject, 40.3% had Stage II and 27% had Stage III or IV injuries.
The Agency for Healthcare Research and Quality (AHRQ) has identified several basic principles for PI prevention: (a) use a validated tool to assess risk such as the Braden Scale and Norton Scale; (b) implement a preventive plan for residents at risk, which should focus on avoiding friction and sheer trauma to at-risk skin regions, as well as an individualized plan to reduce pressure, such as frequent repositioning; and (c) daily inspection of the skin for high-risk residents, as deep tissue damage can occur in as little as two hours. The most common method used to detect early pressure injuries is a visual assessment by a professional caregiver focusing on areas where PIs most frequently develop. This visual assessment is subjective, unreliable, untimely and ineffective as PIs develop under the skin before becoming visible to the naked eye. Technology-based methods for detecting and monitoring PIs have been developed, but none have succeeded in providing an effective solution. These include ulcer detection based on skin conductivity which has relatively low resolution and is influenced by different topical skin conditions (e.g., moisture, urine or feces). Other system solution methods such as electronic medical record programs, which prompt providers to document results of PI screening every shift or day, are of great importance in diagnosing PIs early and preventing progression. Pads designed to specifically cover pressure points such as the sacrum and heels, as well as foam pads designed to wrap around at-risk body parts, are common products. However, it is important to note that some pads can actually be detrimental; for example, supports with cut-outs can have increased pressure at their edges. Hospital-acquired PI rates are increasing while all other hospital-acquired conditions are decreasing (AHRQ, 2019).
PressureSafe
Over the past six years, we have been designing and developing PressureSafe, a novel device that has the potential to provide a reliable method of monitoring and recording patients, providing additional information to healthcare providers as to where and when a pressure injury may occur. The technology platform is designed to record information relating to each patient. The core technologies underlying the PressureSafe device are patent protected (US Patent No. US 10,709,365 and US Patent No. US10,772,541). Our technology is based on the fact that tissues of the human body absorb and reflect the light that surrounds it in different wave lengths (from the UV through visual light to infra-red light), and the light is reflected and scattered back from inside the body through the skin. During this process, the reflected and scattered light through a damaged area changes its properties in comparison to light reflection and scattering from normal healthy tissue. The PressureSafe device is being designed to capture, analyze and identify tissue status to make early PI diagnoses using Spectrographic Analysis, while AI learning software is planned to improve diagnostic accuracy. The PressureSafe device will illuminate the skin with miniature LEDs for a few seconds. The emitted light photons from the device will be absorbed, scattered and reflected back. The device will then measure the absorption and reflectance, and using algorithms, will process the signals to identify and diagnose the scanned area.
As all person’s skin properties are unique, the diagnosing physician must calibrate the device to the specific patient’s skin, a process that takes merely a few seconds and allows personalized diagnosis, improving diagnostic process effectiveness, as the PressureSafe device is designed to measure regardless of skin color. Our technology is being developed to enable the assessment of different subdermal layers by scanning through these skin layers, thus improving the identification of the damage, assessing the subdermal damaged tissue volume and assisting with additional information to allow better treatment efficacy. Measuring the differences of subdermal fluid content and other bio-signals has been developed to detect early formation of PIs and to “raise a flag” to allow the caregivers intervene and prevent their appearance. The bio-signals that our algorithm detects occur in the early inflammatory process, as soon as local subcutaneous tissue function is disturbed, and cells begin to be damaged.
19 Ferrell, B., Josephson, K., Norvid, P. and Alcorn, H., 2000. Pressure Ulcers Among Patients Admitted to Home Care. Journal of the American Geriatrics Society, 48(9), pp.1042-1047.
| 67 |
PressureSafe is a hand-held scanner designed to provide additional information as a decision support system (DSS), to support the care giver effectively with the main diagnostic ability to identify PIs and to differentiate between deep tissue PIs (before they become visible) and Stage 1 PIs. Deep tissue PIs are serious, hospital-acquired deep PIs that form under intact skin, spread in deep tissues and eventually present themselves as full thickness wounds. The PressureSafe is composed of: (a) a handheld optic probe device, which utilizes harmless infra-red light that is placed for a few seconds on suspected areas for performing measurements; (b) a disposable probe tip component, changed between patients to avoid cross-contamination; (c) a software component containing machine learning algorithm for analyzing the collected data; and (d) software for connectivity and downloading the collected data and measurements results to the EMR/EHR systems used by the medical center or homecare company.
PressureSafe is a non-invasive real-time optical monitoring device to support early intervention in PI treatment prior to skin breakage. The device performs a reflectance spectroscopy scan to generate information for the decision maker, while collecting data of subdermal physiological changes together with other bio-signals typical of early formation of PIs in the three skin layers, thus detecting the appearance of life risking pressure injuries. PressureSafe is designed to detect changes at the subepidermal layers of the skin, regardless of skin tone, by measuring differences of subepidermal fluid content and other biomarkers. As soon as local subepidermal tissue function is disturbed and cells begin to disintegrate by pressure exerted upon the body area, our scanner is designed to be able to detect this very early inflammatory process. The technology will allow patient monitoring and immediate reading in a non-invasive way. It has the potential to help to reduce the number of PIs dramatically through early detection, making it attractive for public and private healthcare systems worldwide.
PressureSafe Studies
Our product candidates are in various stages of development. The PressureSafe device is in an advanced stage of development and is planned to be our first go-to-market product.
We have completed the development of the first generation PressureSafe prototype in the second quarter of 2022. In June 2022, IR. Med Ltd., our wholly owned subsidiary, entered into a study agreement with Beit Rivka, a large geriatric hospital in Israel associated with Clalit, the largest Health Maintenance Organization (HMO) in Israel, to conduct a usability study of PressureSafe.
On July 17, 2023, we published our interim report of usability study performed in Israel in leading medical centers with the following results: PressureSafe demonstrated very high efficacy in noninvasively detecting the presence and absence of PIs below the skin’s surface. PressureSafe accurately detected the presence of a PI in 96% of cases. In addition, PressureSafe correctly determined that no wound was present in 91% of cases. The study was conducted at two medical centers owned by Clalit, the world’s second largest health maintenance organization (HMO) and the largest in Israel, namely Beit Rivka Hospital and Rabin Medical Center both in Petah Tikva, where 370 PressureSafe scans were performed on 25 patients who had Stage 1 PIs or deep tissue injuries. No device related safety issues were reported in the total of 44 patients evaluated for safety.
On September 26, 2023, we announced that we signed a Clinical Trial Agreement with the Methodist Healthcare System of San Antonio to conduct a useability study titled “Safety and Efficacy of the PressureSafe Device for Early Detection of Pressure Injury in People with Various Skin Tones, Including Dark Skin Tones.” Methodist Healthcare is recognized as the most respected healthcare provider in its region. With a network of 85 hospitals, 9 of which are acute care facilities, Methodist Healthcare employs more than 11,000 people, including 2,700 physicians. Approximately 50% of subjects recruited for the upcoming study will have a dark skin tone, thus producing comparative data on PressureSafe’s accuracy as a decision support device in detecting early-stage PIs in people of darker and lighter skin tones. Early-stage PIs can be more difficult to see on dark skin tones with the current standard of care for the detection of PIs, which is visual skin inspection.
On February 20, 2024, we reported 92% efficacy for PressureSafe. Data from the study conducted at two medical centers owned by Clalit, the world’s second largest HMO and the largest in Israel, Beit Rivka Hospital and Rabin Medical Center, presented at the NPIAP 2024 Annual Conference on February 16 and 17, 2024 in San Antonio, Texas. The 14-day efficacy portion of the single arm, bi-center study evaluated 38 patients at high risk of pressure injury development. A total of 924 scans were conducted on 154 body locations. Nurses conducting the scans were blinded to PressureSafe’s results, which were encrypted. PressureSafe detected Stage 1 pressure injuries with 92% sensitivity and 88% specificity. Additional portions of the study evaluated safety, as well as device calibration and validation. Total data from 66 patients was obtained for safety analysis and no safety signals were identified in 1,493 scans. Based on these data, the study concluded that PressureSafe is a safe, efficient, and valuable method for early detection of pressure injuries.
| 68 |
We are currently working on completing the development of the commercial version of the PressureSafe device, planned to be launched during September 2024, following the listing with the FDA in April 2024.
DiaSafe Market Potential
We are now in the development stages of the Software/Hardware, algorithms and optics to allow early detection of incipient DFU in the lower limbs. An adjustment to the PressureSafe proven technology allows us to reduce the development period and approach the relevant markets faster. We plan to initiate a clinical study in Clalit, Israel’s largest HMO, to train the developed algorithm and test patients. Following the Israeli trial, we aim to conduct an American based trial, potentially during the first half of 2025.
DFU Market Potential
About 5% of patients with diabetes mellitus develop foot ulcers, and approximately 1% end up with an amputation.
The lifetime risk of a person with diabetes developing a foot ulcer ranges between 19% to 34%. These ulcers are the leading cause of non-traumatic amputations in the U.S. and are responsible for more hospital admissions than any other diabetic complication.
Our goal over the next five years is to establish our technology and related products as the gold standard for the targeted sectors. The key elements of our strategy are as follows:
Develop and expand a balanced and diverse pipeline of products and product candidates. Our core platform technologies will include innovative spectrographic analysis tools for diagnostic aid, AI, devices and product candidates in various development and clinical stages. We plan to add products and product candidates to our pipeline by expanding our technologies being developed to additional indications and through investing in new technologies, products and product candidates. By maintaining this multi-product approach, we aim to provide a broad and comprehensive product offering, which we believe will result in multiple value inflection events, reduced risks to our potential business associated with a particular product or product candidate and increased return on investment. Furthermore, product candidates that we develop may create attractive collaboration opportunities with pharma, diagnostics, medical devices and medical supplies companies.
Target large and growing patient populations with significant unmet medical needs. PIs and ear infections are medical conditions afflicting large and growing global patient populations, each with significant unmet medical needs such as requiring earlier and more accurate diagnosis, reducing the widespread reliance on antibiotics and optimizing the delivery of medical services, thereby improving the efficacy and safety of treatment.
Maintain a global, diverse network of specialists to accelerate knowledge synergies and innovation. We plan to utilize a global network of specialists to identify large and growing patient populations with significant unmet medical needs, evaluate and prioritize potential technologies, assist in designing development plans and diagnostic protocols and determine potential indications of our platform technologies to our target patient populations in various territories. We believe that maintaining this diverse network of industry specialists will allow us to continue to maximize knowledge and cost synergies, utilize shared commercial infrastructure across products, reduce risks of development and commercialization delays to our overall business and leverage our current and future platform technologies and technologies for additional products and product candidates.
Establish distribution channels to maximize the commercial potential of our products. We plan to seek out collaborative arrangements with major healthcare providers to facilitate market adoption of our product candidates. In this respect, we have entered into a distribution agreement covering the United States. See below “Distribution Agreement”. We believe that such institutions are well-positioned to directly benefit from improvements in accurate diagnosis and reduction of cost of care associated with the use of our product candidates. We also believe that the marginal cost of our product candidates compared to potential savings will make it economical for healthcare institutions to adopt our products, regardless of whether or not additional costs of purchase of these products will be covered by third-party payors, such as government healthcare programs and commercial insurance companies. Through cooperation with healthcare providers, we aim to develop and prove an economic model beneficial to them. Thereafter, we plan to engage with private insurance plans to develop reimbursement programs encouraging the use of our product candidates. We expect that adoption rates of our product candidates will increase if hospitals and other medical institutions are compensated, in full or in part, for additional costs incurred when purchasing our products.
| 69 |
Disposable unit/Pay Per Use (PPU) business model - Our developing business model will be based on disposable need to be changed per each patient examined. This will allow potential customers to pay only per use of the device, with minimal investing in equipment, and have great potential to generate substantial revenues to the company. This is especially important following the COVID-19 pandemic, where cross contamination of any kind is forbidden. As we develop large, accumulated databases, we plan to offer software as a service (SAAS) to our customers.
R&D and New Product Development
We believe our strong research and development (R&D) capabilities are one of our principal competitive strengths. Our R&D activities are conducted at our subsidiary’s facility in Israel. Our team of employees and sub-contractors is comprised of current and future dedicated research and development employees, system architects, algorithm developer engineers, software engineers, electronics and electro-optics engineers, quality engineers and regulatory and health experts, who are responsible for the R&D, development and testing of our technologies and product candidates.
We plan to increase our R&D team as necessary to meet our product development goals and milestones and deliver the products in the right time to market and in the required quality.
Distribution Agreement
On October 7, 2022, we entered into an exclusive Distribution and License Agreement, or the Distribution Agreement, with PI Prevention Care LLC, a Delaware limited liability company, or the Distributor, pursuant to which the Distributor received exclusive royalty bearing rights to promote, market and sell solely in the United States our PressureSafe monitoring device.
The Distributor is a recently formed Delaware entity comprised of persons and other entities including Company shareholders, who are active in the markets relating to senior care facilities, hospitals, home care centers, hospital equipment distributors, among others, throughout the United States and who are familiar with and have wide experience in addressing and responding to the needs of these medical care organizations.
Under the Distribution Agreement, the Distributor is solely responsible for the distribution, marketing and sales of the PressureSafe and its accompanying components and agreed undertake all commercially reasonable efforts to establish the necessary distribution and sales network for the Products by not later than the date on which the Company shall have received all regulatory and other clearance required to launch the commercialization of the PressureSafe Solution (such Date being the “Commercial Launch Date”). Prior to the Commercial Launch Date, the Distributor is to invest such resources as is reasonable such that upon the occurrence of the Commercial Launch Date there will be a commercially reasonable distribution network in place for the immediate marketing of the Product. On June 18, 2024, we provided the Distributor with a notice of the Commercial Launch Date being September 20, 2024.
The Distribution Agreement provides for the payment of annual licensing fees. The Distribution Agreement also specifies the prices of each component of the Products payable to the Company and also provides for minimum annual purchase requirements of Product components in order to maintain exclusivity. If for whatever reason the Distributor does not comply with the minimum purchase requirements in any year, the Distributor can continue to have a non-exclusive license and distribution rights in the United States if the Distributor pays the annual license fee.
Subject to the compliance by the Distributor of its obligation under the Distribution Agreement, including the purchase by the Distributor of minimum annual purchase requirements of the components of the Products, the Distribution Agreement continues in effect for a term of 13 years following the Commercial Launch Date. At the end of the initial three- and eight-year periods, the parties are to enter into good faith negotiations as to the pricing of the Products and the minimum purchase quantities for the subsequent period. The Distributor also agreed not to distribute any products that compete with the Products.
| 70 |
Intellectual Property
General
We rely on a combination of patents, trade secrets, non-disclosure agreements and other intellectual property to protect the proprietary technologies that we believe are important to our business. Our success will depend in part on our ability to obtain and maintain patent and other proprietary protection for commercially important inventions and know-how, defend and enforce our patents, maintain our licenses, preserve our trade secrets and operate without infringing valid and enforceable patents and other proprietary rights of third parties. We also rely on continuing technological innovation to develop, strengthen and maintain our proprietary position in the field of diagnostic decision support software devices.
The IR based core technologies underlying the PressureSafe device are covered by patent issued (US Patent No. US 10,709,365 and US Patent No. US10,772,541) issued on July 14, 2020, and September 15, 2020, respectively. All our patents are marked under “system and method for noninvasive analysis of subcutaneous tissue”. Such patents are owned by IR. Med Ltd. and are valid through August 2034.
These patents are based on physical phenomena of light reflection from the surface of the skin. The PressureSafe device irradiates the surface of tissue with harmless infrared and visual light radiation. The reflected light from the tissue changes its physical properties according to the level of injury in the sub dermal tissue (under the skin). Comparing the reflected light from a healthy tissue and reflected light from a suspected injured tissue allows early detection of sub dermal PIs.
During 2022, we applied for two new provisional patents in the US. and European Union, one for the PressureSafe device and one for Nobiotics.
During 2023, we applied for two patents in the U.S. protecting different configurations of our devices and new technological developments. We also applied for two design patents covering our unique design of our devices and accessories.
During 2023, we were granted one patent from the Israeli Patent Office supporting the American patents already approved.
As of the date of this report, a significant portion of our granted U.S. patent applications and pending patent applications in foreign jurisdictions is directed to enhance both the PressureSafe device and other future applications. However, some of these patent applications may not result in issued patents, and not all issued patents may be maintained in force for their entire term.
Competition
We operate in highly competitive segments of the medical device markets. We face competition from many different sources, including commercial medical device enterprises, academic institutions, government agencies and private and public research institutions. Many of our competitors have significantly greater financial, product development, manufacturing and marketing resources than us. Large medical device companies have extensive experience in clinical testing and obtaining regulatory approval for medical devices. We also may compete with these organizations to recruit scientists and clinical development personnel. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies.
We expect PressureSafe, if and when commercially available, to compete directly with Bruin’s Biometrics Provizio SEM Scanner, which is currently commercially available in the U.S., U.K. and the E.U. As we intend with the PressureSafe device, the Bruin scanner is marketed to senior care facilities, as well as other health care centers. Bruin’s product is based on electro-resistance measurement of the skin’s moisture, a method that is significantly different from the approach contained in the PressureSafe device, which utilizes real-time optical monitoring device combined with AI-based capabilities for pre-emptive detection of PIs in different settings. In addition, new developments, including the development of other medical device technologies and methods of preventing PIs, occur in the medical device industry at a rapid pace.
| 71 |
Diabetic foot ulcer assessment is a process composed of collecting a patient’s background data and point of care measurements, such as lower limbs data (pulse quality, level of neuropathy, etc.). The collected information allows rough assessment of a DFU to develop. Since there is no way to measure direct biomarkers associated with DFU, we are developing the technology and a device that will measure direct biomarkers demonstrating development of subdermal DFU.
Manufacturing
We do not own or operate manufacturing facilities. While we plan to depend on third-party contract manufacturers for device manufacturing, we plan to perform the final assembly, quality control and release of finished goods in our facilities.
Manufacturers of our products will be required, among other things, to comply with applicable FDA/EMA manufacturing requirements contained in the FDA/EMA’s Quality System Regulation (QSR). The QSR requires manufacturing quality assurance and quality control as well as the corresponding maintenance of records and documentation.
Major changes to the device generally require regulatory approval before being implemented (e.g., adding new indications and additional labeling claims, etc.).
Under FDA Medical Device Reporting (MDR) regulations, medical device manufacturers are required to report to the FDA information that a device has or may have caused or contributed to a death or serious injury or has malfunctioned in a way that would likely cause or contribute to death or serious injury if the malfunction of the device or one of our similar devices were to recur. Discovery of problems with a product after product release may result in restriction on a product or manufacturer, including withdrawal of the product from the market.
We do not have any current contractual relationships for the manufacture of commercial supplies of any of our product candidates. We intend to enter into contract manufacturing agreements and one or more back-up manufacturers for the commercial production of our product candidates in the near future.
Distribution and Revenue Generation
We intend to establish sales and marketing structures and strategic partnerships in the United States, U.K. and Europe to support all of our product candidates.
The target market for our PressureSafe device is relevant healthcare settings (i.e., hospitals, senior care facilities, homecare companies, etc.), nursing homes and a growing segment of long-term homecare givers. Towards that end, in the third quarter of 2022, we began preparations in anticipation of commercialization of PressureSafe in the United States during 2024, pending regulatory approvals, which have been delayed. A distribution agreement was entered into with PI Prevention Care LLC, a newly formed entity focused on marketing to the senior care facility, hospital and homecare markets. The Distributor, which received exclusive rights for PressureSafe distribution across the United States, includes personnel who have many years’ experiences in addressing and responding to the needs of these types of organizations. Under the terms of the Agreement which were publicly disclosed, to maintain exclusivity, the Distributor is obligated to comply with minimum purchase requirements of the device and accompanying disposables.
Once we receive the appropriate sales approvals, we expect the marketing will be done with local partners who have the relevant abilities and connections in each territory where the company will ask to sell the products. Since each country has its own specific healthcare system, a local partner (one or more) will be chosen to address the specific market needs in terms of regulation, technical support, etc. Pricing will be determined by the local partner, taking into account all overhead expected costs, regulation requirements and reimbursement methods.
| 72 |
The DiaSafe once developed, will be marketed pending our receipt of the appropriate sales approvals. We expect the marketing will be done with local partners who have the relevant abilities and connections per each relevant distribution territory. Since each country has its own specific healthcare system, a local partner (one or more) will be chosen to address the specific market needs in terms of regulations and technical support. Pricing will be determined by the local partner, taking into account all overhead expected costs, regulation requirements and reimbursement methods.
In both the PressureSafe and the DiaSafe devices, the revenue stream is expected to be generated mainly from the disposables and PressureSafe solution as a service (PSaaS) that are needed for the proper operation of the device, while the device itself likely be given under lease agreements. It is envisioned that the disposable component will be mass produced.
It is expected that market penetration will be achieved through original equipment manufacturing agreements with one of several large medical device companies already selling to the target market. At the current time, we have no commitments from any such distributors or original equipment manufacturing partners.
Facilities
Our subsidiary occupies approximately 130 square meters of facilities located in Rosh Pinna industrial zone, Israel, under an agreement for shared office space and services that expires upon 90 days’ notice by either our subsidiary or the landlord. Through December 31, 2023, we were paying a monthly rent of 24,750 New Israeli Shekels, or NIS, per month (approximately $6,780).
Legal Proceedings
On May 29, 2023, a lawsuit was filed against us, the Subsidiary and Mr. Aharon Klein, or the Plaintiff, one of our Directors and our Chief Technology Officer in the Tel Aviv District Court of Israel by an individual who provided, on apart-time basis, certain consulting services to the Subsidiary between October 2015 through October 2016, prior to the acquisition of the Subsidiary by us. The lawsuit alleges breach of contract by the defendants based on non-payment of amounts purportedly owed to the Plaintiff in respect of the services rendered, including the market value of our common stock that the Plaintiff alleges should have been issued to him in respect of his services. The suit seeks declaratory judgment that the defendants breached certain agreements with the Plaintiff and claimed damages in the aggregate amount of approximately $2.1 million based on the current exchange rate between the U.S. Dollar and the Israeli NIS. We believe that the allegations are baseless and without merit. We intend to vigorously defend our rights.
Other than as set forth above, the Company is not currently involved in any legal proceedings. However, from time to time we may become involved in various legal proceedings that arise in the ordinary course of business, including actions related to our intellectual property. Although the outcomes of these legal proceedings cannot be predicted with certainty, we are currently not aware of any such legal proceedings that arise in the ordinary course of business, including actions related to our intellectual property. Although the outcomes of these legal proceedings cannot be predicted with certainty, we are currently not aware of any such legal proceedings or claims that we believe, either individually or in the aggregate, will have a material adverse effect on our business, financial condition or results of operations.
We are also subject to the U.S. Foreign Corrupt Practices Act (“FCPA”), which prohibits improper payments or offers of payments to foreign governments and their officials for the purpose of obtaining or retaining business and requires companies to maintain accurate books and records and a system of internal accounting controls. Safeguards we implement to discourage improper payments or offers of payments by our employees, consultants, and others may be ineffective, and violations of the FCPA and similar laws may result in severe criminal or civil sanctions, or other liabilities or proceedings against us, any of which would likely harm our reputation, business, financial condition and result of operations.
| 73 |
Employees & Consultants
As of June 24, 2024, we had 6 employees and 1 service provider, on a full-time basis, with 3 on a part time basis, engaged in product research and development at IR. Med Ltd.
During February 2024, as a result of financial difficulties, we notified 10 of our employees, including our then-Chief Executive Officer, of the termination of their employment. The effective termination dates vary based on contractual notice periods, which range between March 22, 2024, and April 22, 2024. During March 2024 following the approval of the IIA plan, we canceled the termination notice of five employees.
Israeli labor laws govern the length of the workday, minimum wages for employees, procedures for hiring and dismissing employees, determination of severance pay, annual leave, sick days, advance notice of termination of employment, equal opportunity and anti-discrimination laws and other conditions of employment. Subject to specified exceptions, Israeli law generally requires severance pay upon the retirement, death or dismissal of an employee, and requires us and our employees to make payments to the National Insurance Institute, which is similar to the U.S. Social Security Administration. Our employees have defined benefit pension plans that comply with the applicable Israeli legal requirements. Our employees are not represented by a labor union. We consider our relationship with our employees to be good. To date, we have not experienced any work stoppages.
Government Regulation and Product Approval
The healthcare industry in which we operate is highly regulated, and the services we provide are subject to a complex set of healthcare laws and regulations. We and our customers must comply with a variety of requirements, including among others, HIPAA, HITECH, regulations issued by the Department of Health and Human Services and the Centers for Medicare and Medicaid Services, a number of fraud and abuse laws, such as the federal Anti-Kickback Statute and the False Claims Act, and comparable state laws. We have structured our operations to comply with these laws and other regulatory and contractual requirements.
Government authorities in the United States, at the federal, state and local levels, as well as other countries, extensively regulate, among other things, the research, development, testing, manufacture, quality control, approval, labeling, packaging, storage, record-keeping, promotion, advertising, distribution, post-approval monitoring and reporting, marketing and export and import of products such as those we are developing.
We may also be subject to various federal laws targeting fraud and abuse in the healthcare industry. For example, the federal Anti-Kickback Statute prohibits, among other things, any person from knowingly or willfully offering, soliciting, receiving or paying remuneration (a term interpreted broadly to include anything of value, including, for example, gifts, discounts and credits), directly or indirectly, in cash or in kind, to induce or reward, or in return for, either the referral of an individual for, or the purchase, order or recommendation of, an item or reimbursable, in whole or in part, under a federal healthcare program such as the Medicare and Medicaid programs. The federal Anti-Kickback Statute has been broadly interpreted by federal courts and agencies, and potentially subject many healthcare business arrangements to government investigation, enforcement, and prosecution, which can be costly and time consuming. In addition, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation.
In addition, the False Claims Act, or FCA, prohibits anyone from, among other things, knowingly presenting or causing to be presented, for payment to federal programs (including Medicare and Medicaid) claims for items or services that are false or fraudulent. Although we would not submit claims directly to payors, we could be held liable under the False Claims Act if we are deemed to “cause” the submission of false or fraudulent claims by, for example, including inaccurate information in draft medical notes for physicians, or if our documentation services are found to have caused clinicians to have inaccurately attested to “Meaningful Use” criteria. Claims for services that were induced by kickbacks and in violation of the federal Anti-Kickback Statute may also form the basis for FCA liability. In recent years, many cases have been brought against healthcare companies by the government and by “whistleblowers,” which have resulted in judgments and settlements involving substantial payments to the government by the companies involved. Penalties for a violation of the FCA include fines for each false claim, plus up to three times the amount of damages caused by each false claim. The cost to defend against allegations can also be substantial.
| 74 |
HIPAA also established federal criminal statutes that prohibit, among other things, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, including private third-party payers, and knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. Similar to the Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation.
Additionally, many of the states in which we operate also have similar fraud and abuse focused laws that apply to our business. The scope of these laws and the interpretations of them vary from state to state and are enforced by state courts and regulatory authorities, each with broad discretion. Some state fraud and abuse laws apply to items or services reimbursed by any payor, including patients and commercial insurers, not just those reimbursed by a federally funded healthcare program.
Violations of these laws are punishable by substantial penalties and other remedies, including monetary fines, civil penalties, administrative penalties, criminal sanctions (in the case of Anti-Kickback Statute), exclusion from participation in FHCPs, forfeiture of amounts collected in violation of such laws and additional reporting requirements and compliance oversight obligations. Similarly, state anti-kickback and false claims laws may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by any source, not only government programs.
Government Regulations
Before we can market our product candidates to the public in the U.S., we believe the products will need to obtain clearance for commercial sales. Our devices will be subject to ongoing regulation by the FDA in the U.S. and other federal, state, and local regulatory bodies.
FDA regulations govern, among other things, product design and development, manufacturing, labeling, pre-clinical and clinical trials, post-market adverse event reporting, post-market surveillance, complaint handling, repair or recall of products, product storage, record keeping, pre-market clearance, advertising and promotion and sales and distribution.
Unless an exemption applies, each medical device, such as our PressureSafe, DiaSafe and Nobiotics that are intended to be commercially distributed in the United States, requires 510(k) clearance from the FDA. Based on the FDA guidance documents that we have reviewed, we expect to be subject to the shorter and more streamlined 510(k) process for PressureSafe, which typically involves less risk of uncertainty and the submission of less supporting documentation, without the costly clinical trials (though of course no prior guarantee can be provided as to such regulatory treatment). Generally, gaining 510(k) clearance for a product depends on demonstrating that the subject product is “substantially equivalent” to a previously cleared 510(k) device.
On April 9, 2024, the PressureSafe decision support device received an FDA listing certification. PressureSafe is classified as a Class I device. We are currently working on completing the development of the commercial version of the PressureSafe device, planned to be launched during September 2024, following the listing under the FDA. We intend to pursue approximately the same regulatory track for the DiaSafe device.
Failure to comply with applicable regulatory requirements can result in enforcement actions by the FDA and other regulatory agencies, which may include any of the following sanctions: untitled letters or warning letters, fines, injunctions, consent decrees, civil or criminal penalties, recall or seizure of our current or future products, operating restrictions, partial suspension or total shutdown of production, refusal of or delay in granting 510(k) clearance of new products or modified products or rescission of previously granted 510(k) clearances. Any of these sanctions could result in higher than anticipated costs and have a material adverse effect on our reputation, business and financial condition. See “Risk Factor - Government Regulation,” above.
| 75 |
The FDA can delay, limit or deny clearance of our proposed devices for many reasons, including:
| ● | our inability to demonstrate that our product is safe and effective for its intended users; | |
| ● | our inability to demonstrate that our product is the “substantial equivalent” of a previously cleared device; | |
| ● | the data from clinical studies that we undertake may be insufficient to support clearance; and | |
| ● | failure of the manufacturing process or facilities we use to meet applicable requirements. |
In addition, the FDA may change its pre-market policies, adopt additional regulations, revise existing regulations or take other actions which may prevent or delay clearance of our devices.
Any delay in or failure to receive or maintain regulatory compliance prior to marketing our devices could prevent us from generating revenue therefrom or achieving profitability.
Additionally, the FDA and other regulatory authorities have broad enforcement powers. Regulatory enforcement or inquiries or other increased scrutiny on us could dissuade some customers from using our proposed product and adversely affect our reputation and the perceived safety and efficacy of our proposed devices. If the FDA requires us to go through a more rigorous examination for our proposed product than we currently expect, such as requiring additional testing further verification or other procedures, we may require substantial additional funding sooner than anticipated commercialization of the DiaSafe device may be severely delayed. Being subject to an extended period of scrutiny or being required to conduct expensive clinical trials would be particularly harmful to our business because our proposed devices currently constitute our only products.
Ongoing Regulation by FDA.
Placing the PressureSafe and DiaSafe devices on the market requires in addition (depending on the stage of approval of each device):
| ● | Establishment registration and device listing; | |
| ● | Quality system regulation, which requires manufacturers, including third-party manufacturers, to follow stringent design, testing, control, documentation and other quality assurance procedures during all aspects of the manufacturing process; | |
| ● | Labeling regulations and FDA prohibitions against the promotion of products for uncleared, unapproved or “off-label” uses, and other requirements related to advertising and promotional activities; | |
| ● | Medical device reporting (MDR) regulations, which require that manufacturers report to the FDA if their device may have caused or contributed to a death or serious injury or malfunctioned in a way that would likely cause or contribute to a death or serious injury if the malfunction were to recur; | |
| ● | Corrections and removals reporting regulations, which require that manufacturers report to the FDA field corrections and product recalls or removals if undertaken to reduce a risk to health posed by the device or to remedy a violation of the 1938 Federal Food, Drug, and Cosmetic Act (FDCA), that may present a risk to health; | |
| ● | Labelling and Unique Device Identification (UDI) regulations; and | |
| ● | Post-market surveillance regulations, which apply when necessary to protect the public health or to provide additional safety and effectiveness data for the device. (Refer to the section below) |
Post-Approval Requirements
Although premarket clinical trials provide important information on a device’s safety and effectiveness, it is possible that new safety concerns will emerge once the device is on the market. As a result, the FDA continues to monitor device performance after a device has been approved. FDA officials conduct routine inspections of medical device manufacturing facilities across the United States. Manufactures may be informed of inspections in advance, or the inspections may be unannounced. Inspections may be routine or caused by a particular problem. The purpose of these inspections is to make sure developers are following good manufacturing practices. Furthermore, the FDA can shut down a manufacturing facility if required standards are not met.
Usability Studies
We have completed the development of the first generation PressureSafe prototype in the second quarter of 2022. In June 2022, IR. Med Ltd., our wholly owned subsidiary, entered into a study agreement with Beit Rivka, a large geriatric hospital in Israel associated with Clalit, the largest Health Maintenance Organization (HMO) in Israel, to conduct a usability study of PressureSafe.
| 76 |
In February 2023, our subsidiary IR. Med Ltd. entered into an agreement with Rabin Medical Center in Israel to perform a usability study, as an additional study center to the current study that we have been performing at Beit-Rivka, a large geriatric hospital in Israel. The agreement is to conduct a usability study of our proprietary and patent protected “PressureSafe” device, which we plan to launch as a decision support system (DSS) tool for care givers in hospitals, nursing homes and homecare companies.
On July 17, 2023, we published our interim report of usability study performed in Israel in leading medical centers with the following results: PressureSafe demonstrated very high efficacy in noninvasively detecting the presence and absence of PIs below the skin’s surface. PressureSafe accurately detected the presence of a PI in 96% of cases. In addition, PressureSafe correctly determined that no wound was present in 91% of cases. The study was conducted at two medical centers owned by Clalit, the world’s second largest health maintenance organization (HMO) and the largest in Israel, namely Beit Rivka Hospital and Rabin Medical Center, both in Petah Tikva, where 370 PressureSafe scans were performed on 25 patients who had Stage 1 PIs or deep tissue injuries. No device related safety issues were reported in the total of 44 patients evaluated for safety.
On September 26, 2023, we announced that we signed a Clinical Trial Agreement with the Methodist Healthcare System of San Antonio to conduct a useability study titled “Safety and Efficacy of the PressureSafe Device for Early Detection of Pressure Injury in People with Various Skin Tones, Including Dark Skin Tones.” Methodist Healthcare is recognized as the most respected healthcare provider in its region. With a network of 85 hospitals, 9 of which are acute care facilities, Methodist Healthcare employs more than 11,000 people, including 2,700 physicians. Approximately 50% of subjects recruited for the upcoming study will have dark skin tone, thus producing comparative data on PressureSafe’s accuracy as a decision support device in detecting early-stage PIs in people of darker and lighter skin tones. Early-stage PIs can be more difficult to see on dark skin tones using the current standard of care for the detection of PIs, which is visual skin inspection.
On February 20, 2024, we announced highly favorable proof of efficacy data for PressureSafe. Data from the study conducted at two medical centers owned by Clalit, Beit Rivka Hospital and Rabin Medical Center, presented at the NPIAP 2024 Annual Conference on February 16 and 17, 2024 in San Antonio, Texas. Dr. Gal Maydan of Beit Rivka Hospital Geriatric Rehabilitation Center, and Principal Investigator of the study, presented the data in a poster titled “Near Infra-Red Spectroscopy for early detection of stage 1 pressure injury and deep tissue injury – clinical study results”. While the current standard of care for the detection of pressure injuries is visual and tactile clinical evaluation, physiological changes below the skin’s surface, including inflammation and interstitial fluids precede changes on the surface. The objective of the study was to evaluate the sensitivity, specificity, and usability of PressureSafe to detect early-stage pressure injuries Stage 1 suspected deep tissue injuries (sDTI) before skin breakage, compared to standard of care. PressureSafe detected biomarkers and changes in tissue structures under the skin’s surface as they relate to pressure injuries.
The 14-day efficacy portion of the single arm, bi-center study evaluated 38 patients at high risk of pressure injury development. A total of 924 scans were conducted on 154 body locations. Nurses conducting the scans were blinded to PressureSafe’s results, which were encrypted. PressureSafe detected Stage 1 pressure injuries with 92% sensitivity and 88% specificity. Additional portions of the study evaluated safety, as well as device calibration and validation. Total data from 66 patients was obtained for safety analysis and no safety signals were identified in 1,493 scans. Based on these data, the study concluded that PressureSafe is a safe, efficient, and valuable method for early detection of pressure injuries.
In addition, we plan to conduct usability studies in the U.S. or other countries on the PressureSafe and on the DiaSafe. Additional regulations govern the approval, initiation, conduct, documentation and reporting of clinical studies to regulatory agencies in the countries or regions in which they are conducted. Such investigational use is generally also regulated by local and institutional requirements and policies which usually include review by an ethics committee or institutional review board (IRB). Failure to comply with all regulations governing such studies could subject the company to significant enforcement actions and sanctions, including halting of the study, seizure of investigational devices or data, sanctions against investigators, civil or criminal penalties and other actions. Without the data from one or more clinical studies, it may not be possible for us to secure the data necessary to support certain regulatory submissions, to secure reimbursement or demonstrate other requirements. We cannot assure that access to clinical investigators, sites and subjects, documentation and data will be available on the terms and timeframes necessary.
| 77 |
Reimbursement
Our current go-to-market strategy does not contemplate or rely upon governmental or third-party payor reimbursement. However, in the future we plan to seek reimbursement for product candidates as a means to expand the adoption of products and broaden our customer base.
To the extent that we adopt a market strategy which is in whole or in part reliant on third-party reimbursement, commercial sales of our future products will depend in part on the availability of reimbursement from such third-party payors, including government health administrative authorities, managed care providers, private health insurers and other organizations. Each third-party payor may have its own policy regarding which products it will cover, the conditions under which it will cover such products, and how much it will pay for such products. Third-party payors are increasingly examining the medical necessity and cost effectiveness of medical products and services in addition to safety and efficacy and, accordingly, significant uncertainty exists as to the reimbursement status of newly approved devices. Further, healthcare policy and payment reform models and medical cost containment models are being considered and/or adopted in the United States and other countries. Legislative and/or administrative reforms to applicable reimbursement systems may significantly reduce reimbursement for the services in which our products are used or result in the denial of coverage for such services outright. As a result, third-party reimbursement adequate to enable us to realize an appropriate return on our investment in research and product development may not be available for our products.
Other Healthcare Laws and Compliance Requirements
In the United States, our activities are potentially subject to regulation by various federal, state and local authorities in addition to the FDA, including the Centers for Medicare and Medicaid Services (formerly the Health Care Financing Administration), other divisions of the United States Department of Health and Human Services (e.g., the Office of Inspector General), the United States Department of Justice and individual United States Attorney offices within the Department of Justice, and state and local governments.
Anti-Kickback Statutes in the United States
The U.S. federal anti-kickback statute prohibits persons from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in exchange for or to induce either the referral of an individual, or the furnishing, arranging for or recommending of a good or service, for which payment may be made in whole or in part under a U.S. federal healthcare program such as the Medicare and Medicaid programs. The definition of “remuneration” has been broadly interpreted to include anything of value, including gifts, discounts, the furnishing of supplies or equipment, payments of cash and waivers of payments. Several courts have interpreted the statute’s intent requirement to mean that, if any one purpose of an arrangement involving remuneration is to induce referrals or otherwise generate business involving goods or services reimbursed in whole or in part under U.S. federal healthcare programs, the statute has been violated. Penalties for violations include criminal penalties and civil sanctions such as fines, imprisonment and possible exclusion from Medicare, Medicaid and other U.S. federal healthcare programs. The reach of the federal anti-kickback statute was broadened by the Affordable Care Act (ACA), which, among other things, amends the intent requirement of the federal anti-kickback statute. Pursuant to the statutory amendment, a person or entity no longer needs to have actual knowledge of this statute or specific intent to violate it in order to have committed a violation. The ACA further provides that the government may assert that a claim including items or services resulting from a violation of the federal anti-kickback statute constitutes a false or fraudulent claim for purposes of the U.S. False Claims Act or the Civil Monetary Penalties statute, which imposes penalties against any person who is determined to have presented or caused to be presented a claim to a federal health program that the person knows or should know is for an item or service that was not provided as claimed or is false or fraudulent.
| 78 |
The U.S. federal anti-kickback statute is broad and prohibits many arrangements and practices that are lawful in businesses outside of the healthcare industry. Recognizing that the statute is broad and may technically prohibit many innocuous or beneficial arrangements, the Office of Inspector General (OIG) of the Department of Health and Human Services, has issued a series of regulations, known as “safe harbors.” These safe harbors set forth provisions that, if all their applicable requirements are met, will assure healthcare providers and other parties that they will not be prosecuted under the anti-kickback statute. The failure of a transaction or arrangement to fit precisely within one or more safe harbors does not necessarily mean that it is illegal or that prosecution will be pursued. However, conduct and business arrangements that do not fully satisfy an applicable safe harbor may result in increased scrutiny by government enforcement authorities such as the OIG or the U.S. Department of Justice.
Many states have adopted laws similar to the U.S. federal anti-kickback statute. Some of these state prohibitions are broader than the U.S. federal statute and apply to the referral of patients and recommendations for healthcare items or services reimbursed by any source, not only the Medicare and Medicaid programs. Government officials have focused certain enforcement efforts on marketing of healthcare items and services, among other activities, and have brought cases against individuals or entities with sales personnel who allegedly offered unlawful inducements to potential or existing physician users in an attempt to procure their business.
U.S. Health Insurance Portability and Accountability Act of 1996 (HIPAA)
HIPAA imposes criminal and civil liability for executing a scheme to defraud any healthcare benefit program, including private payors, or making false statements relating to healthcare matters. HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act, or HITECH, which governs the conduct of certain electronic healthcare transactions and protects the security and privacy of protected health information, can impose civil or criminal liability for violations of its provisions.
In addition, we may be subject to data privacy and security regulation by both the federal government and the states in which we conduct our business. HIPAA, as amended by HITECH and its implementing regulations, imposes certain requirements relating to the privacy, security and transmission of individually identifiable health information. Among other things, HITECH makes HIPAA’s privacy and security standards directly applicable to “business associates”- independent contractors or agents of covered entities that receive or obtain protected health information in connection with providing a service on behalf of a covered entity. HITECH also increased the civil and criminal penalties that may be imposed against covered entities, business associates and possibly other persons, and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce the federal HIPAA laws and seek attorney’s fees and costs associated with pursuing federal civil actions. In addition, state laws govern the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts.
International Regulation
The European Commission is the legislative body responsible for the EU MDR (Medical Device Regulation) with which manufacturers selling medical products in the European Union and the European Economic Area, or EEA, must comply. The European Union has adopted regulations of the design, manufacture, labeling, clinical studies, post-market clinical follow-up, post-market surveillance and vigilance for medical devices. Devices that comply with the requirements of a relevant EU MDR will be entitled to bear the CE conformity marking, indicating that the device conforms to the essential requirements of the applicable regulations and, accordingly, can be marketed throughout the European Union and EEA, after being certified by a Notified Body. The centralized procedure provides for the grant of a single marketing authorization that is valid for all European Union member states.
In addition to regulations in the United States, there are a variety of foreign regulations governing clinical trials and commercial sales and distribution of any product candidates. The approval process varies from country to country, and the time may be longer or shorter than that required for bringing the product to the U.S. market.
Corporate Information
IR-Med, Inc. was incorporated in the state of Nevada on April 20, 2007, under the name “Monster Motors, Inc.” On June 24, 2009, the corporate name was changed to Eco2 Forests, Inc. During September 2012, Eco2 Forests, Inc., accepted a court ordered receiver who authorized a reduction of the authorized shares from 900,000,000 to 500,000,000 and in November 2012 effectuated a 16,000 to 1 stock split. In February 2013, the Company underwent a change of control. On March 25, 2013, Eco2 Forests, Inc., effectuated a 4 to 1 reverse stock split in addition to changing the corporate name to International Display Advertising, Inc.
Available Information. The Company’s reports, including Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K and other information filed with, or furnished to, the SEC, including amendments to such reports, are available on the SEC’s website that contains such reports, proxy and information statements, and other information regarding companies that file electronically with the Commission. This information is available at www.sec.gov. In addition, all the Company’s public filings can be accessed through the Company’s website at https://www.ir-medical.com/investors/#Financial_statements.
| 79 |
MANAGEMENT
The following table sets forth information regarding our executive officers and directors as of the date of this prospectus:
| Name | Age | Positions | ||
| Oded Bashan | 77 | Chairperson of the Board of Directors | ||
| Aharon Klein | 63 | Interim Chief Executive Officer, Chief Technology Officer, Director | ||
| Sharon Levkoviz | 50 | Chief Financial Officer | ||
| Aharon Binur | 60 | Chief Development Officer | ||
| Yaniv Cohen | 44 | Director | ||
| Ohad Bashan | 53 | Director | ||
| Ron Mayron | 60 | Director | ||
| Avital Rosenberg | 49 | Director |
Business Experience
The following is a brief account of the education and business experience of our current directors and executive officers:
Oded Bashan, co-founded IR. Med Ltd. with Aharon Klein and, since September 2013 has been serving as Chairman of IR. Med Ltd. Upon the effectiveness of our acquisition in December 2020 of IR. Med Ltd. (the “Acquisition”), he was appointed to Board of Directors and on January 20, 2021, was appointed as Chairman of the Board, and on April 6, 2021, he was appointed Chief Executive Officer on an interim basis following the resignation of Ms. Davidson Mund. Mr. Bashan has over 35 years of experience in managing, building and running technology companies. He was Founder, CEO & chairman of OTI Ltd. from 1990 to 2013, a Nasdaq traded global technology leader with more than 250 employees. Since January 2013, together with his son Mr. Ohad Bashan, they have been managing several private companies engaged in biotech. Previously, Mr. Bashan served as the president of Electro-Galil form 1984 to1990. He was awarded the Leading Businessman Award in Management, Business and Economics by the Israeli Institute of Public Opinion. Mr. Bashan holds both B.Sc. and M.Sc. in Economics and Business management from the Hebrew University of Jerusalem.
Aharon Klein was appointed as Interim Chief Executive Officer effective February 28, 2024. Mr. Klein co-founded IR. Med Ltd. in September 2013 and has served as a director of the Company since. He also served as the Company’s Chief Operating Officer from September 2013 until December 2020. Since December 2020, Mr. Klein serves as the Company’s Chief Technology Officer. Mr. Klein is a medical device and biotech expert, with a strong clinical background. Prior to founding the Company, from 2004 to 2007 Mr. Klein co-founded and served as Chief Executive Officer of Fertiligent, a start-up company focused on innovative fertility treatments, which was acquired by a United Kingdom based investment group in 2008. From 2008 to 2013, immediately prior to co-founding of the Company, he founded a medical device company developing infrared based diagnostic tools for diagnosing colon cancer without the need for biopsies (optical biopsies). Mr. Klein graduated from the Faculty of Engineering in the Technion - Israel Institute of Technology. Mr. Klein is experienced in initiating and running medical device start-up companies, including development, clinical trials and regulatory affairs.
| 80 |
Sharon Levkoviz. Mr. Levkoviz was appointed to Chief Financial Officer upon the effectiveness of the Acquisition. Mr. Levkoviz served from 2011-2021 in Achdut Israel Ltd., an Israeli company providing accounting and economic consulting services, as regional manager. Prior to that period, Mr. Levkoviz served as a Chief Controller at OTI, Nasdaq traded company, from 2005 through 2011. Mr. Levkoviz received his CPA from Ramat Gan College and a B.A. in Business Administration from Rupin College in Israel. In addition, Mr. Levkoviz served 10 years as a chairman of finance and human resource committee at Ohalo College. He also served five years as a director at the development company of Katzrin and eight years as a member of Katzrin plenum.
Aharon Binur. Mr. Aharon Binur was appointed as Chief Development Officer on April 29, 2021, to lead product development. Mr. Binur is an electronics engineer who graduated from the Technion in Haifa, Israel. He was an electronics engineer at OTI from November 1999 through February 2001 and became a development manager at a subsidiary of OTI in March 2001 and held several other positions at OTI through April 2013. Mr. Binur also served as CTO (August 2009 through November 2014) and VP of R&D (March 2017 through April 2021) at Lehavot, an advanced fire protection system company. Mr. Binur has extensive experience in multidisciplinary technological management, including software, hardware and mechanics, development of final systems and products for clients, while maintaining high quality and international standards. Mr. Binur has a unique and creative approach to technology management, including patents registered in his name.
Dr. Yaniv Cohen. (Ph. D, M.Sc.) Mr. Yaniv Cohen co-founded IR. Med Ltd. in September 2013 and has served as the R&D manager. Following the completion of the acquisition of IR. Med Ltd. by our Company (the “Acquisition”), he was appointed as a Director of the Board and the Company’s Chief Scientific Officer. Dr. Cohen acts as a scientist at the Weizmann Institute of Science, he is a skilled researcher, engineer and entrepreneur, with years of experience leading R&D development for medical device companies. His fields of expertise include electro-optics, infrared spectroscopy and medical devices products using infrared light. His current research at Prof. Avigdor Scherz’s lab at the Weizmann Institute of Science is focused on the launch of Immune Photoactivated Cancer Therapy treatments for solid tumors using Near Infrared lasers combined with drug therapy. He has successfully led the development of medical devices from early concept to commercial product. Previous work experience includes positions with Cisco and Tokyo Electron Israel. Dr. Cohen earned his Ph.D. from the renowned National Research University HSE, School of Electronic Engineering Institute of Electronics and Mathematics (MIEM HSE) in Moscow. Russia (2022).
Ohad Bashan, Mr. Bashan joined the Board upon the completion of the Acquisition. Mr. Bashan is an entrepreneur, innovator and executive with a proven track record of more than 25 years of building, leading and running technology companies from startup to NASDAQ trade. He is an experienced director, after having served on the boards of private and publicly traded companies in the U.S., Israel, China, Poland and France. From 1998 to 2013, Mr. Bashan held several senior positions at OTI, which was acquired by Nyax Ltd. From 2013 to the present, Mr. Bashan has run a management services business. Mr. Bashan holds a B.A. in business from the College of Business Management, Tel Aviv, with specializations in marketing and finance, and an M.B.A. from Pepperdine University, California.
Ron Mayron. Mr. Mayron has extensive experience in the pharmaceutical and medical equipment industries and has held various, significant senior management positions, both local and global, within Teva Pharmaceutical Industries Ltd. (“Teva”) over the last 21 years. During his career at Teva, Mr. Mayron served in various VP positions. Most recently he was CEO of Teva Israel and VP Israel and Africa from June 2009 until September 2013. Mr. Mayron’s core expertise is in marketing, sales and distribution, mergers and acquisitions, business development, global operation and supply chain and strategic development. Mr. Mayron currently serves on the board of directors of Innocan Pharma Corporation (CSE: INNO), Nurexone Biologic Inc. (TSX: NRX), IceCure Medical Ltd. (NASDAQ: ICCM), BioLight Life Sciences Ltd. (TASE: BOLT), Entera Bio (NASDAQ: ENTX) and Kadimastem Ltd. (TASE: KDST). Mr. Mayron holds a B.Sc. in Industrial Engineering & Management from Ben Gurion University and M.B.A from Tel-Aviv University.
Avital Rosenberg. Ms. Avital Rosenberg, is an international business-oriented counsel and an Israeli licensed attorney with over 20 years of extensive experience in ESG, Compliance, M&A, Securities, and complex business development international transactions. Ms. Rosenberg currently serves as the Executive Vice President and Chief Legal Officer at Rafael Advanced Defense Systems Ltd. (“Rafael”), a position she has held since 2019. In this role, she is responsible for all legal affairs, compliance, and regulatory activities of the Rafael Group worldwide. Ms. Rosenberg is also a member of Rafael’s Corporate Senior Management, General Counsel of the board of directors, and head of the legal department since 2019. Ms. Rosenberg served as Deputy Chief Legal Officer and Head of the Business Development & International Transactions Unit at Rafael, from 2008 to 2019, leading mergers and acquisition transactions in over 12 countries and specializing in the U.S. market. She also held the role of Corporate Secretary and board member in more than 25 of Rafael’s subsidiaries, and managed major international defense transactions and public financing activities. Ms. Rosenberg received her LL.M in Commercial Law and her Executive MBA from Bar-Ilan University.
| 81 |
Family Relationships
Oded Bashan is the father of Ohad Bashan.
Director Independence
Our Board of Directors has reviewed the materiality of any relationship that each of our directors has with us, either directly or indirectly. Based on this review, our Board of Directors has determined that Ron Mayron and Avital Rosenberg are “independent directors” as defined in Nasdaq Listing Rules and Rule 10A-3 promulgated under the Exchange Act. The Company intends to change the composition of the Board of Directors in order to meet the rules of the NASDAQ Capital Market, and will include seven (7) board members, with four (4) board members meeting the independence requirements contemplated by Rule 10A-3 under the Exchange Act.
Corporate Governance
We strongly believe that our success depends on all of our employees identifying with our company’s purpose and understanding how their work contributes to the Company’s overall strategy. To this end, we engaged in an inclusive all-company process to develop our company purpose, vision, mission and values.
Our corporate culture and values, along with our employees are our most valuable. These values, are:
| ● | Passion, | |
| ● | Integrity, | |
| ● | Excellence, | |
| ● | Responsibility, | |
| ● | Innovation, and | |
| ● | Spirit of Collaboration. |
These values form part of our goal setting and review process to ensure accountability to these values at all levels. In order to further ensure that we live our values, and our culture stays unique and strong, our Board of Directors and executive management team put significant focus on our human capital resources.
We utilize a variety of channels to facilitate open and direct communication, including: (i) monthly all-hands staff meetings, (ii) regular open learning forums to promote peer learning or town hall meetings with executives; (iii) regular ongoing update communications; and (iv) employee surveys beyond the annual engagement survey referenced above on an as-needed basis.
Term of Office of Directors
We currently have authorized six directors. In accordance with our Amended and Restated Articles of Incorporation and Amended and Restated Bylaws, our Board of Directors is divided into three classes with staggered three-year terms. At each annual meeting of stockholders commencing with the meeting in 2021, the successors to the directors whose terms then expire will be elected to serve until the third annual meeting following the election. Our directors are divided among the three classes as follows:
| ● | the Class I director are Yaniv Cohen and Avital Rosenberg. Their terms will expire at the annual meeting of stockholders to be held in 2025; | |
| ● | the Class II directors are Ohad Bashan and Ron Mayron, and their terms will expire at the annual meeting of stockholders to be held in 2026; and | |
| ● | the Class III directors are Oded Bashan and Aharon Klein, and their terms will expire at the annual meeting of stockholders to be held in 2024. |
| 82 |
On February 18, 2024, Ms. Inna Martin notified the Board of Directors of her resignation from the Board, effective immediately. The resignation of Ms. Martin as a director was not related to any disagreement with the Company on any matter relating to the Company’s operations, policies or practices.
On March 27, 2024, Mr. Yoram Drucker notified the Board of Directors of his resignation from the Board, effective immediately. The resignation of Mr. Drucker as a director was due to the current composition of the Board.
Committees of the Board of Directors
Our Board of Directors has established an audit committee which operates under a charter that has been approved by our board.
Our Board of Directors has determined that all of the members of our audit committee are independent as defined under the rules of the NASDAQ Capital Market. In addition, all members of the audit committee meet the independence requirements contemplated by Rule 10A-3 under the Exchange Act. We currently have a board member that qualifies as an “audit committee financial expert” as defined in Item 407(D)(5) of Regulation S-K.
We currently do not have a nominating or compensation committees or committees performing similar functions, nor does our Company have a written nominating or compensation charter. Our directors believe that it is not necessary to have such committees at this time because the director(s) can adequately perform the functions of such committees.
Audit Committee
Our Audit Committee is comprised of Mr. Ron Mayron and Ms. Avital Rosenberg, both independent directors. Mr. Mayron is the Chairman of the Audit Committee and is an “audit committee financial expert” as defined in Item 407(d)(5)(ii) of Regulation S-K. Prior to the consummation of this offering, we intend to add additional members to our Audit Committee.
| 83 |
The audit committee’s main function is to oversee our accounting and financial reporting processes and the audits of our financial statements. This committee’s responsibilities include, among other things:
| ● | appointing our independent registered public accounting firm; | |
| ● | evaluating the qualifications, independence and performance of our independent registered public accounting firm; | |
| ● | approving the audit and non-audit services to be performed by our independent registered public accounting firm; | |
| ● | reviewing the design, implementation, adequacy and effectiveness of our internal accounting controls and our critical accounting policies; | |
| ● | discussing with management and the independent registered public accounting firm the results of our annual audit and the review of our quarterly unaudited financial statements; | |
| ● | reviewing, overseeing and monitoring the integrity of our financial statements and our compliance with legal and regulatory requirements as they relate to financial statements or accounting matters; | |
| ● | reviewing on a periodic basis, or as appropriate, any investment policy and recommending to our board any changes to such investment policy; | |
| ● | preparing the report that the SEC requires in our annual proxy statement; | |
| ● | reviewing and approving any related party transactions and reviewing and monitoring compliance with our code of conduct and ethics; and | |
| ● | reviewing and evaluating, at least annually, the performance of the audit committee and its members including compliance of the audit committee with its charter. |
Compensation Committee. During 2023, we did not, and do not currently, have a compensation committee. If and when we satisfy the other initial listing standards for listing our common stock on Nasdaq or another national exchange, we intend to establish a compensation committee of the Board of Directors.
Nominating Committee. During 2023, we did not, and do not currently, have a nominating committee. If and when we satisfy the other initial listing standards for listing our common stock on Nasdaq or another national exchange, we intend to establish a nominating committee of the Board of Directors.
| 84 |
EXECUTIVE AND DIRECTOR COMPENSATION
The following table summarizes the compensation earned in each of our fiscal years that ended December 31, 2023 and 2022 by our named executive officers, which consists of our chief executive officer and our two next most highly compensated executive officers who earned more than $100,000 during the fiscal year ended December 31, 2023 and 2022 and were serving as executive officers as of such date and our former chief executive officers. We refer to the executive officers listed below as the Named Executive Officers.
Summary Compensation Table
| Name and Principal Position | Year | Salary ($) | Bonus ($) | Option Awards ($) (1) | All other compensation ($) (2) | Total ($) | ||||||||||||||||
| Moshe Gerber, | 2023 | 88,337 | 342,996 | 43,062 | 474,395 | |||||||||||||||||
| Chief Executive Officer (3) | 2022 | 92,896 | 195,074 | 40,776 | 328,746 | |||||||||||||||||
| Oded Bashan, | 2023 | - | 273,728 | - | 273,728 | |||||||||||||||||
| Executive Chairman | 2022 | 95,163 | 299,730 | - | 394,893 | |||||||||||||||||
| Aharon Klein, | 2023 | 90,657 | - | 10,417 | 101,074 | |||||||||||||||||
| Interim Chief Executive Officer and Chief Technology Officer | 2022 | 124,954 | 3,442 | 16,500 | 144,896 | |||||||||||||||||
| Aharon Binur- | 2023 | 114,558 | 92,268 | 45,845 | 252,671 | |||||||||||||||||
| Chief Development Officer | 2022 | 126,528 | 48,553 | 51,180 | 226,262 | |||||||||||||||||
| Dr. Rom Eliaz, former | 2023 | - | - | - | - | |||||||||||||||||
| Chief Executive Officer (4) | 2022 | 111,038 | 7,795 | 51,321 | 170,153 | |||||||||||||||||
Mr. Tzur Di Cori, former Chief Executive Officer (5) | 2023 | 31,135 | 48,031 | 14,811 | 93,977 | |||||||||||||||||
| 1. | In accordance with SEC rules, the amounts in this column reflect the fair value on the grant date of the option awards granted to the named executive, calculated in accordance with ASC Topic 718. Stock options were valued using the Black-Scholes model. The grant-date fair value does not necessarily reflect the value of shares which may be received in the future with respect to these awards. The grant-date fair value of the stock options in this column is a non-cash expense for us that reflects the fair value of the stock options on the grant date and therefore does not affect our cash balance. The fair value of the stock options will likely vary from the actual value the holder receives because the actual value depends on the number of options exercised and the market price of our Common Stock on the date of exercise. For a discussion of the assumptions made in the valuation of the stock options, see Note 9.C to the Annual Report on Form 10-K for the year ended December 31, 2023. |
| 2. | For 2023 and 2022, represents the compensation as described under the caption “All Other Compensation” below. |
| 3. | Mr. Gerber was appointed Chief Executive Officer on May 2, 2022, and resigned as Chief Executive Officer on May 21, 2023.
|
| 4. | Dr. Eliaz was appointed Chief Executive Officer on June 22, 2021, and resigned as Chief Executive Officer on August 31, 2022. |
| 5. | Mr. Tzur Di-Cori was appointed Chief Executive Officer on October 15, 2023, and on February 22, 2024, the Board of Directors terminated his employment. |
| 85 |
All Other Compensation
The following table provides information regarding each component of compensation for fiscal years 2023 and 2022 included in the “All Other Compensation” column in the “Summary Compensation Table” above. Represents amounts paid in New Israeli Shekels (NIS) and converted at average exchange rates for the year.
| Name | Year | Automobile and Related Expenses $ (1) | Social Benefits $ (2) | Total $ | ||||||||||
| Moshe Gerber | 2023 | 17,267 | 25,795 | 43,062 | ||||||||||
| 2022 | 11,794 | 28,982 | 40,776 | |||||||||||
| Aharon Klein | 2023 | 10,417 | - | 10,417 | ||||||||||
| 2022 | 16,500 | - | 16,500 | |||||||||||
| Aharon Binur | 2023 | 15,805 | 30,040 | 45,845 | ||||||||||
| 2022 | 17,859 | 33,321 | 51,180 | |||||||||||
| Dr. Rom Eliaz | 2023 | - | - | - | ||||||||||
| 2022 | 19,936 | 31,385 | 51,321 | |||||||||||
| Tzur Di Cori | 2023 | 5,755 | 9,056 | 14,811 | ||||||||||
| 1. | Represents a leased automobile expense. |
| 2. | These are comprised of contributions by us to savings, health, severance, pension, disability and insurance plans generally provided in Israel, including health, education, managerial insurance funds, and redeemed vacation pay. This amount represents Israeli severance fund payments, managerial insurance funds, disability insurance, supplemental education fund contribution and social securities. See discussion below under “Narrative Disclosure to Summary Compensation Table.” |
Outstanding Equity Awards on December 31, 2023
| Name | Grant Date | Number of Securities Underlying Options (#) Exercisable | Number of Securities Underlying Options (#) Unexercisable | Option Exercise Price ($) | Option Expiration Date | |||||||||||
| Moshe Gerber | May 2, 2022 | 433,333 | - | 0.32 | December 31, 3030 | |||||||||||
| Oded Bashan | June 20,2021 | 240,000 | - | 0.32 | * | |||||||||||
| Oded Bashan | December 30, 2022 | 1,200,000 | - | 0.58 | * | |||||||||||
| Aharon Klein | June 20,2021 | 240,000 | - | 0.32 | * | |||||||||||
| Aharon Binur | June 20,2021 | 165,000 | 135,000 | 0.32 | * | |||||||||||
| Aharon Binur | November 14.2022 | 125,000 | 175,000 | 0.58 | * | |||||||||||
| Dr. Rom Eliaz | June 20,2021 | 150,000 | - | 0.32 | July 31, 2027 | |||||||||||
| Tzur Di Cori | September 27, 2023 | - | 1,000,000 | 0.58 | * | |||||||||||
| 86 |
The following table sets forth information concerning equity awards held by each of our Named Executive Officers as of December 31, 2023.
| * | Options expiration date is ten (10) years from vesting |
Narrative Disclosure to Summary Compensation Table
Our Board follows the following processes and procedures for the consideration and determination of executive and director compensation:
In establishing compensation amounts for executives, we seek to provide compensation that is competitive in light of current market conditions and industry practices. Accordingly, we will generally review market data, which is comprised of proxy-disclosed data from peer companies and information from nationally recognized published surveys for the biopharmaceutical industry, adjusted for size. The market data helps the committee gain perspective on the compensation levels and practices at the peer companies and to assess the relative competitiveness of the compensation paid to our executives. The market data thus guides us in its efforts to set executive compensation levels and program targets at competitive levels for comparable roles in the marketplace. We then consider other factors, such as the importance of each executive officer’s role to the Company, individual expertise, experience, performance, retention concerns and relevant compensation trends in the marketplace, in making its final compensation determinations.
Elements of Compensation
In addition to each officer’s base salary, our executive officer compensation program consists of a cash incentive bonus plan and discretionary stock option awards in addition to customary benefits. The amounts of compensation awarded for each element of the Company’s compensation program (i.e., base salary, bonuses and stock options) are reviewed in connection with the Company’s performance.
Base Salary
Annual base salaries compensate our executive officers for fulfilling the requirements of their respective positions and provide them with a level of cash income predictability and stability with respect to a portion of their total compensation. We believe that the level of an executive officer’s base salary should reflect the executive’s performance, experience and breadth of responsibilities, our understanding of salaries for similar positions within our industry and any other factors relevant to that particular job.
Base salaries are typically negotiated at the outset of an executive’s employment. Salary levels are considered annually as part of our performance review process, but also in cases including promotion or other changes in the job responsibilities of an executive officer. For named executive officers, initial base salaries generally are established in connection with negotiation of an offer of employment and employment agreement. Increases in base salary have several elements. In addition to promotion and increased responsibilities, merit and Company-wide general increases are also taken into consideration.
Stock-Based Awards
Historically, we have generally granted stock options to our employees, including our named executive officers, in connection with their initial employment with us. We also have historically granted stock options on an annual basis as part of annual performance reviews of our employees.
| 87 |
Our equity award program is the primary vehicle for offering long-term incentives to our executives. We do not have any equity ownership guidelines for our executives, which is consistent with other pre-commercial biotechnology companies that use stock options as the long-term incentive vehicle. Further, we believe that equity grants provide our executives with a strong link to our long-term performance, create an ownership culture and help to align the interests of our executives and our stockholders. In addition, the vesting feature of our equity awards contributes to executive retention by providing an incentive for our executives to remain in our employment during the vesting period. We expect that our Board will continue to use annual equity awards to compensate our executive officers. We may also make additional discretionary grants, typically in connection with the promotion of an employee, to reward an employee, for retention purposes or in other circumstances as the Board deems appropriate.
Employment and Severance Arrangements
We consider it essential to the best interests of our stockholders to foster the continuous employment of our key management personnel. In this regard, we recognize that the possibility of a change in control may exist and that the uncertainty and questions that it may raise among management could result in the departure or distraction of management personnel to the detriment of the Company and our stockholders. In order to reinforce and encourage the continued attention and dedication of certain key members of management, we have entered into written employment agreements with certain of our named executive officers that, while at-will, contain certain change in control and severance provisions.
Employment Agreements
Moshe Gerber. The Company’s subsidiary IR. Med Ltd. (“IR. Med Ltd.”) entered into an Employment Agreement (the “Agreement”) setting forth the terms of his employment and compensation. Under the Agreement, Mr. Gerber is entitled to an annual salary of the current New Israeli Shekel equivalent of approximately $136,752, payable on monthly basis. Mr. Gerber is also provided with a leased automobile. Under the Agreement, Mr. Gerber is also entitled to the following: (i) Manager’s Insurance under Israeli law to which IR. Med Ltd. contributes amounts equal to (a) 8-1/3 percent for severance payments, and 6.5%, or up to 7.5% (including disability insurance) designated for premium payment (and Mr. Gerber contributes an additional 6%) of each monthly salary payment, and (b) 7.5% of his salary (with Mr. Gerber contributing an additional 2.5%) to an education fund, a form of deferred compensation program established under Israeli law. If Mr. Gerber’s employment is terminated by us without cause on or prior to the first anniversary of his employment, then we must pay Mr. Gerber (a) the accrued obligations earned through the date of termination, (b) a lump-sum payment of an amount equal to two month of his base salary at the time of his termination; if such termination occurs after such date, then we must pay to Mr. Gerber (a) the accrued obligations earned through the date of termination, (b) a lump-sum payment of an amount equal to three months of his base salary at the time of his termination. Under the Agreement, Mr. Gerber was awarded options under the Company’s employee stock option plan for 1,300,000 shares of the Company’s common stock at a per share price of $0.32, vesting as follows: 25% of the option shares (i.e., 325,000 options shares) vest on the first anniversary of employment and the balance in six bi-annual instalments of 162,500 shares, beginning the first instalment on the bi-annual period ending October 31, 2023 and thereafter at the end of each subsequent six months, provided that Mr. Gerber is then in our employ.
The agreement contains (i) customary confidentiality obligations which are not limited by the term of the agreement, (ii) certain non-compete provisions during the term of the agreement and 12 months thereafter and (iii) certain non-solicitation provisions during the term of the agreement and for one year thereafter.
On May 21, 2023, the Company and Mr. Moshe Gerber, executed and delivered a termination and settlement agreement pursuant to which Mr. Gerber resigned from his role as Chief Executive Officer of the Companies. Mr. Gerber’s resignation was not associated with or attributable to any disagreement with the Company or the Company’s independent auditor, including without limitation, any matter relating to the Company’s accounting principles or practices, financial statement disclosures, internal controls, management or operations.
Pursuant to the terms of the termination and settlement agreement, Mr. Gerber was available to the Company until August 19, 2023. Mr. Gerber continued to receive his salary and social benefits under his employment agreement with the Company through August 2023. In addition, the Company agreed that Mr. Gerber was entitled to accelerated vesting of certain of his options under the Company’s 2020 Incentive Stock Option Plan; therefore, Mr. Gerber was entitled to purchase an aggregate of 433,333 shares of the Company’s common stock through December 31, 2030, at the exercise price per share of $0.32, which reflects the exercise price in the original grant. Additionally, subject to his compliance with the terms of the termination and settlement agreement, on August 19, 2023, Mr. Gerber received an additional 21,000 NIS (approximately $5,780 at the current exchange rate).
| 88 |
Oded Bashan. On June 27, 2022, our subsidiary IR. Med Ltd. and Bashanti Ltd., a company that is owned by Oded Bashan, entered into consulting agreement. The Agreement provides for a continuous term and may be terminated by either party at any time with at least 12 months prior written notice. Pursuant to this agreement, Mr. Bashan’s annual fee compensation is $144,000 plus value added tax (VAT). On October 26, 2022, Mr. Bashan agreed to defer payment of his fee pending a capital raise. On December 12, 2022, Mr. Bashan was awarded options under the Company’s employee stock option plan for 1,200,000 shares of the Company’s common stock at a per share price of $0.58, of which 600,000 were vested upon grant and the balance vest on December 30, 2023, subject to his continued service. The options are exercisable through the tenth anniversary of the grant.
Aharon Klein. On December 24, 2020, our subsidiary IR. Med Ltd. and Mr. Klein entered into an amended and restated consulting agreement (the “Klein Service Agreement”) replacing a service agreement dated October 1, 2019, between IR. Med Ltd. and the Company). The Klein Service Agreement provides for a continuous term and may be terminated by either party at any time, provided that if Mr. Klein resigns, he shall provide at least 30 days’ prior written notice. Pursuance to this agreement, Mr. Klein’s annual fee compensation was increased to $144,000 plus VAT. On October 26, 2022, Mr. Klein agreed to defer 50% of his payment of his fee pending a capital raise. On October 19 ,2023 the board of directors of the company decided to approve the payment pursuant to his original service agreement commencing October 2023. In addition, Mr. Klein is eligible to receive an automobile allowance of $750 per month. If Mr. Klein’s employment is terminated (i) by us without cause or (ii) by him for any, then we must pay Mr. Klein (a) the accrued obligations earned through the date of termination and (b) a lump-sum payment of an amount equal to one month of his base salary at the time of his termination.
The agreement contains (i) customary confidentiality obligations which are not limited by the term of the agreement, (ii) certain non-compete provisions during the term of the agreement and 12 months thereafter and (iii) certain non-solicitation provisions during the term of the agreement and for one year thereafter. Mr. Klein also agreed to assign certain intellectual property rights to IR. Med Ltd.
Aharon Binur. On March 2, 2021, IR. Med Ltd. and Aharon Binur entered into an employment agreement pursuant to which Mr. Binur oversees the development of our product candidates which are in various stages of development. Under the agreement with Mr. Binur, he is paid an annual salary of the current New Israeli Shekel equivalent of approximately $120,170, payable on monthly basis. IR. Med Ltd. is authorized to terminate the employment agreement for any reason subject to payment of two months’ salary. Under the terms of the employment agreement with him, Mr. Binur also receives Manager’s Insurance under Israeli law for his to which IR. Med Ltd. contributes amounts equal to (a) 8-1/3 percent for severance payments, and 6.5%, or up to 7.5% (including disability insurance) designated for premium payment (and Mr. Binur contributes an additional 6%) of each monthly salary and (b) 7.5% of his salary (with Mr. Binur contributing an additional 2.5%) to an education fund, a form of deferred compensation program established under Israeli law. Mr. Binur is also provided with a leased automobile. On June 20, 2021, Mr. Binur was awarded options under the Company’s employee stock option plan for 300,000 shares of the Company’s common stock at a per share price of $0.32, of which 15,000 were vested upon grant and the balance vest at the end of each calendar quarter at the rate of 15,000 shares per quarter, beginning with the quarter ended September 31, 2021, subject to his continued employment. The options are exercisable through the tenth anniversary of grant.
On November 14, 2022, Mr. Binur was awarded options under the Company’s employee stock option plan for 300,000 shares of the Company’s common stock at a per share price of $0.58, vesting at the end of each calendar quarter at the rate of 25,000 shares per quarter, beginning with the quarter ended December 31, 2022, subject to his continued employment. The options are exercisable through the tenth anniversary of the grant.
| 89 |
Dr. Rom Eliaz. On June 22, 2021, Dr. Rom Eliaz and IR. Med Ltd. entered into an employment agreement providing for the employment of Dr. Eliaz as Chief Executive Officer (the “Eliaz Employment Agreement”). Under the Eliaz Employment Agreement, Dr. Eliaz is entitled to an annual salary of the current New Israeli Shekel equivalent of approximately $150,427, payable on a monthly basis. Mr. Eliaz is also provided with a leased automobile. Under the Eliaz Employment Agreement, Dr. Eliaz is also entitled to the following: (i) Manager’s Insurance under Israeli law for the benefit of Dr. Rom Eliaz pursuant to which IR. Med Ltd. contributes amounts equal to (a) 8-1/3% for severance payments, and 6.5%, or up to 7.5% (including disability insurance) designated for premium payment (and Dr. Eliaz contributes an additional 6%) of each monthly salary payment, and (b) 7.5% of his salary (with Dr. Eliaz contributing an additional 2.5%) to an education fund, a form of deferred compensation program established under Israeli law. The Eliaz Employment Agreement provides that his annual salary will be increased to the New Israeli Shekel equivalent of approximately $186,000 upon (i) the successful capital raise by the Company of at least $5.0 million in net proceeds, (ii) the successful completion of a prototype of the PressureSafe device, as determined by the Company’s board of directors and (iii) receipt by the Company of a letter of intent for the large scale commercial purchase/order of the PressureSafe device.
On April 25, 2022, Dr. Eliaz resigned from his position. In connection with his resignation, the Company agreed that Dr. Eliaz continued to receive his salary and benefits under his employment agreement through August 31, 2022. In addition, the Company agreed that Dr. Eliaz is entitled to the accelerated vesting of the next instalment of options (previously granted to in June 2021) which vested on June 30, 2022, for an aggregate total of options for 150,000 shares of the Company’s common stock and that such options may be exercised through the original exercise expiration date of July 31, 2027, at an exercise price per share of $0.32.
Tzur Di-Cori. On October 15, 2023, Mr. Di-Cori was appointed to serve as the Company’s Chief Executive Officer. In conjunction with his appointment, the Company and Mr. Di-Cori entered into an employment agreement, pursuant to which he was subject to standard confidentiality, intellectual property assignment and non-compete provisions. In addition, in consideration of his service, Mr. Di-Cori received a monthly gross salary of NIS 45,000 ($11,842 approximately) and options to purchase 1,000,000 shares of the Company’s common stock at an exercise price of $0.58 per share, subject to and in accordance with the terms and conditions of the Company’s 2020 Incentive Stock Plan.
On February 22, 2024, as a result of financial difficulties, the Company notified Mr. Di-Cori, of the termination of his employment.
Potential Payments upon Change of Control or Termination following a Change of Control
Our agreements with our named executive officers provide incremental compensation in the event of termination, as described herein. Generally, we currently do not provide any severance specifically upon a change in control nor do we provide for accelerated vesting upon change in control. Termination of employment also impacts outstanding stock options.
Due to the factors that may affect the amount of any benefits provided upon the events described below, any actual amounts paid or payable may be different than those shown in this table. Factors that could affect these amounts include the basis for the termination, the date the termination event occurs, the base salary of an executive on the date of termination of employment and the price of our common stock when the termination event occurs.
The following table sets forth the compensation that would have been received by each of our executive officers had they been terminated as of December 31, 2023.
| Name | Salary $ | Social benefits $ | Total $ | |||||||||
| Oded Bashan | - | - | - | |||||||||
| Aharon Klein | 13,500 | - | 13,500 | |||||||||
| Aharon Binur | 19,178 | 5,000 | 24,178 | |||||||||
| Tzur Di Cori | 12,162 | 3,734 | 15,896 | |||||||||
| 90 |
Director Compensation
The following table sets forth for each non-employee director that served as a director during the year ended December 31, 2023:
Year Ended December 31, 2023
| Name | Fees Earned or Paid in Cash ($) | Stock Awards ($) | Option Awards ($) (1) | Non-Equity Incentive Plan Compensation ($) | Non-Qualified Deferred Compensation Benefits ($) | All Other compensation ($) | Total ($) |
|||||||||||||||||||||
| Inna Martin | 15,696 | 86,649 | (5) | - | - | - | 102,345 | |||||||||||||||||||||
| Ohad Bashan | - | - | (2) | - | - | - | - | |||||||||||||||||||||
| Ron Mayron | 20,735 | 57,764 | (3) (4) | - | - | - | 178,499 | |||||||||||||||||||||
| 1. | In accordance with SEC rules, the amounts in this column reflect the fair value on the grant date of the option awards granted to the named executive, calculated in accordance with ASC Topic 718. Stock options were valued using the Black-Scholes model. The grant-date fair value does not necessarily reflect the value of shares which may be received in the future with respect to these awards. The grant-date fair value of the stock options in this column is a non-cash expense for us that reflects the fair value of the stock options on the grant date and therefore does not affect our cash balance. The fair value of the stock options will likely vary from the actual value the holder receives because the actual value depends on the number of options exercised and the market price of our common stock on the date of exercise. For a discussion of the assumptions made in the valuation of the stock options, see Note 1. C (Stock Based Compensation) to our financial statements, which are included in our Annual Report on Form 10-K for the year ended December 31, 2023. |
| 2. | In respect of 240,000 options, all of which were vested. |
| 3. | In respect of 240,000 options, all of which were vested. |
| 4. | In respect of 160,000 options, all of which will vest as of September 30, 2024. |
| 5. | In respect of 240,000 options, all of which will vest as of September 30, 2024. |
Compensation Policy for Non-Employee Directors.
In January 2021, the Board of Directors adopted an updated compensation policy for non-employee directors which replaced the previous non-employee director compensation terms, and which became effective January 2021. Under the policy, each director is to receive an annual cash compensation of $5,000 and $1,000 per meeting or $500 for a virtual meeting. On October 26, 2022, the Board of Directors decided to freeze compensation for non-employee directors. On August 13, 2023, the Board of Directors decided to repay the independent directors for fees incurred in the fourth quarter of 2022 and the first half of 2023, and moving forward, to continue paying the independent directors board attendance fee.
| 91 |
CERTAIN RELATIONSHIPS AND RELATED PARTY TRANSACTIONS
In addition to the compensation arrangements discussed in the section entitled “Executive Compensation,” the following is a description of each transaction since June 24, 2022 and each currently proposed transaction in which:
| ● | we have been or are to be a participant; |
| ● | the amounts involved exceeded or will exceed the lesser of $120,000 and 1% of our total assets; and |
| ● | any of our directors, executive officers or holders of more than 5% of our capital stock, or an affiliate or immediate family member of the foregoing persons, had or will have a direct or indirect material interest. |
Other than as described below, there have not been, nor are there any currently proposed, transactions or series of similar transactions to which we have been or will be a party other than compensation arrangements, which are described where required under the section entitled “Executive Compensation.”
In 2015, our subsidiary IR. Med Ltd. received a loan from certain of the former IR-Med stockholders to fund its continuing operations. This loan bore interest at an annual rate ranging in 2023 and 2022, from 2.9%-2.42% annually. The aggregate loan amount was repayable only upon the approval of IR Med’s board of directors and when the Company’s profits reach an amount of NIS 1,500,000 (approximately $414,000 as of December 31, 2023) and upon such terms and such installments as shall be determined by the Company’s board of directors.
In 2017, our subsidiary IR. Med Ltd. received a loan from certain of the former IR-Med stockholder to fund its continuing operations. This loan bears interest at an annual rate ranging from 2.9%-2.42% annually in 2022 and 2023. The aggregate loan amount was repayable only upon the occurrence of an investment round greater than $500,000.
In March 2020, IR. Med Ltd. and the lenders agreed to amend and restate the terms of the above referenced loans (“the Amended loan agreement”) pursuant to which the lender waived all rights to convert their respective outstanding loan amounts, and the repayment date was set to December 31, 2023, or such later date to be agreed between IR. Med Ltd. and the lender. On March 1, 2024, the lenders agreed to extent the repayment date to December 31, 2025. As of December 31, 2023, and 2022 the carrying amounts of these loans were $38 thousand.
On March 6, 2018, certain of IR. Med Ltd.’s shareholders advanced to it a convertible bridge loan in the principal amount of NIS 379,000 ($104,000) (hereinafter, the “2018 CLA”), bearing a per annum interest rate of 3% compounded and accrued annually and, originally payable on December 31, 2018, or a later date agreed to by the then holders of 80% of the outstanding shares of IR. Med Ltd. Under the terms of the 2018 CLA, the loan is convertible by the holders under certain specified circumstances and is automatically convertible upon other terms. In an Exit event (as deveined in the 2018 CLA), the loan is repayable at 200% the outstanding amount or converted, at the option of the majority lenders. In March 2020, the Company and the lenders agreed to amend and restate the 2018 CLA (“the Amended CLA”). According to the Amended CLA, the lenders waived any and all rights to convert their respective outstanding loan amounts, and the repayment date was set to December 31, 2023, or such later date to be agreed by IR. Med Ltd. and the lenders. On March 1, 2024, the lenders agreed to extent the repayment date to December 31, 2025. In addition, in case of an Exit event, as described in the Amended CLA, the loan and all accrued interest will be fully repaid immediately following the Exit event. As of December 31, 2023, and 2022, the carrying amounts of the loans were $123,000 and $124,000, respectively.
For the years ended December 31, 2023, and 2022, the Company paid to two directors and to an entity controlled by a shareholder of the Company an aggregate consideration of US$161 thousand and US$220 thousand, respectively, in respect of research and development services.
For the years ended December 31, 2023, and 2022 the Company paid to one shareholder of the Company and his relative an aggregate consideration of $150 thousand and $111 thousand, respectively in respect consulting services.
For the years ended December 31, 2023, and 2022, the Company paid to four of the Company non-employee directors an aggregate consideration of $36 thousand and $130 thousand respectively in respect of their services.
For the year ended December 31, 2023, the Company paid to four of its officers, salary and related expenses that totaled to $439 thousand. For the year ended December 31, 2022, the Company paid to one director of the Company and four of its officers, salary and related expenses totaled to $651 thousand, respectively, in respect thereof.
For the years ended December 31, 2023, and 2022, the Company paid a yearly amount of $80 thousand and $51 thousand to an entity controlled by two of the Company’s directors for rent and office services, respectively.
Following the adoption of the 2020 incentive stock plan (hereinafter the “Plan”) by the Company on December 23, 2020, and the adoption of the sub plan (the “Israeli appendix”) on April 29, 2021, the Company granted during the years 2023 and 2022 to its directors, officers, and shareholder 1,636,000 and 5,200,000 options to purchase shares of Common Stock respectively.
| 92 |
PRINCIPAL STOCKHOLDERS
The following table sets forth, as of the date of this prospectus, information regarding beneficial ownership of our capital stock by:
| ● | each person, or group of affiliated persons, known by us to beneficially own more than 5% of our common stock; |
| ● | each of our executive officers; |
| ● | each of our directors; and |
| ● | all of our executive officers and directors as a group. |
Beneficial ownership is determined in accordance with the rules of the SEC and includes voting or investment power with respect to the securities. Under the rules of the SEC, a stockholder is deemed to be a beneficial owner of any security of which that stockholder has the right to acquire beneficial ownership in 60 days of June 24, 2024. Except as indicated in footnotes to this table, we believe that the stockholders named in this table have sole voting and investment power with respect to all shares of common stock shown to be beneficially owned by them based on information provided to us by these stockholders. Percentage of ownership is based on 70,699,144 shares of common stock outstanding on June 24, 2024.
| Name and Address of Beneficial Owner | Number of Shares beneficially owned | Percentage Beneficially owned | ||||||
| 5% or more shareholders | ||||||||
| Yaakov Safren | 6,156,120 | (1) | 8.47 | % | ||||
| Paul Coulson | 5,625,000 | (2) | 7.75 | % | ||||
| Yoram Drucker | 4,862,471 | (3) | 6.87 | % | ||||
| Third Eye Investors LLC | 4,687,500 | (4) | 6.49 | % | ||||
| Isamar Margareten | 8,721,307 | (5) | 11.67 | % | ||||
| Liat Electronics Ltd. | 3,850,607 | 5.45 | % | |||||
| Officers and Directors | ||||||||
| Oded Bashan | 10,049,916 | (6) | 13.93 | % | ||||
| Aharon Klein | 8,099,110 | (7) | 11.42 | % | ||||
| Yaniv Cohen | 8,099,136 | (8) | 11.42 | % | ||||
| Ron Mayron | 380,000 | (9) | * | |||||
| Ohad Bashan | 240,000 | (9) | * | |||||
| Aharon Binur | 370,000 | (9) | * | |||||
| Sharon Levkoviz | 390,921 | (9) | * | |||||
| Officers and Directors as a Group (7 persons) | 27,629,083 | 37.34 | % | |||||
| * | less than 1% |
| (1) | Includes 2,006,119 shares issuable upon the exercise of stock options. |
| (2) | Includes 1,875,000 shares issuable under a currently exercisable common stock warrant. |
| (3) | Includes 812,471 shares issuable upon the exercise of stock options. |
| (4) | Yitzchak Rokonsky, of Third Eye Investors LLC (“Third Eye”) has sole voting and dispositive power over shares held by Third Eye. Includes 1,562,500 shares issuable under a currently exercisable common stock warrant. |
| (5) | Includes 4,043,466 shares issuable under a currently exercisable common stock warrant. |
| (6) | Represents (i) 8,609,916 shares owned by Med2Bwell Ltd. (“Med2Bwell”), of which Mr. Bashan has sole voting and dispositive power and (ii) 1,440,000 shares of common stock issuable upon the exercise of stock options. |
| (7) | Includes 240,000 shares issuable upon the exercise of stock options |
| (8) | Includes 240,000 shares issuable upon the exercise of stock options. |
| (9) | Represents shares of common stock issuable upon the exercise of stock options. |
| 93 |
DETERMINATION OF OFFERING PRICE
The offering price has been negotiated between the representatives of the Underwriter and us. In determining the offering price, the following factors were considered:
| ● | prevailing market conditions; |
| ● | our historical performance and capital structure; |
| ● | estimates of our business potential and earnings prospects; |
| ● | an overall assessment of our management; and |
| ● | the consideration of these factors in relation to market valuation of companies in related businesses. |
| 94 |
DESCRIPTION OF THE SECURITIES
Description of Existing Securities
As of June 24, 2024, we had one class of securities registered under Section 12(g) of the Exchange Act: Common Stock. Each of the Company’s securities registered under Section 12(g) of the Exchange Act are quoted on the OTC Market tier, QB. Our common stock is listed on the OTCQB Market under the symbol “IRME.” We intend to apply to list our common stock and warrants on Nasdaq/ under the symbols “IRME.” and “IRMEW”, respectively.
General
As of the date of this prospectus our authorized capital stock consists of 600,000,000 shares of Common Stock. As of June 24, 2024, there were 70,699,144 shares of our common stock outstanding.
In addition, as of the date of this prospectus, we had issued and outstanding:
| ● | options to purchase 13,924,175 shares of our Common Stock, at a weighted average exercise price of $0.42 per share; and | |
| ● | warrants to purchase 12,474,259 shares of our Common Stock, at a weighted average exercise price of $0.89 per share. |
The following summary description of our capital stock is based on the provisions of our certificate of incorporation and bylaws, the applicable provisions of applicable law, including the provision of Chapters 78 and 92A of the Nevada Revised Statutes or NRS.
Common Stock
Each share of Common Stock entitles the holder to one vote on all matters submitted to a vote of the stockholders including the election of directors. Except as otherwise required by law the holders of our Common Stock possess all voting power. According to our bylaws, when a quorum is present or represented at any meeting, the vote of the holders of a majority of the stock having voting power present in person or represented by proxy shall be sufficient to elect members of the Board of Directors or to decide any question brought before such meeting, unless the question is one upon which by express provision of the statutes or of the Certificate of Incorporation, a different vote is required in which case such express provision shall govern and control the decision of such question.. Our bylaws provide those stockholders holding at least a majority of the shares entitled to vote, represented in person or by proxy, constitute a quorum at the meeting of our stockholders. Our bylaws also provide that any action which may be taken by the vote of the stockholders at a meeting may be taken without a meeting if authorized by the written consent of stockholders holding at least a majority of the voting power, unless the provisions of the statutes or of the Articles of Incorporation require a greater proportion of voting power to authorize such action in which case such greater proportion of written consents shall be required.
Our certificate of incorporation and bylaws do not provide for cumulative voting in the election of directors. Because the holders of our common stock do not have cumulative voting rights and directors are generally to be elected by a majority of the votes casts with respect to the directors at any meeting of our stockholders for the election of directors, holders of more than fifty percent, and in some cases less than 50%, of the issued and outstanding shares of our common stock can elect all of our directors.
Provisions of our Restated Certificate of Incorporation and Restated Bylaws
Classified board of directors. Our restated certificate of incorporation and restated bylaws provide that our board of directors is classified into three classes of directors, each with staggered three-year terms. A third party may be discouraged from making a tender offer or otherwise attempting to obtain control of us as it is more difficult and time consuming for stockholders to replace a majority of the directors on a classified board of directors.
| 95 |
Advance Notice Requirements for Stockholder Proposals and Director Nominations. Our restated bylaws provide advance notice procedures for stockholders seeking to bring business before our annual meeting of stockholders or to nominate candidates for election as directors at our annual meeting of stockholders. Our restated bylaws also specify certain requirements regarding the form and content of a stockholder’s notice. These provisions might preclude our stockholders from bringing matters before our annual meeting of stockholders or from making nominations for directors at our annual meeting of stockholders if the proper procedures are not followed. We expect that these provisions might also discourage or deter a potential acquirer from conducting a solicitation of proxies to elect the acquirer’s own slate of directors or otherwise attempting to obtain control of our company.
Amendment of Bylaws. Our bylaws provide that our board of directors may amend our bylaws by a majority vote of our board of directors including any bylaws adopted by our stockholders, but our stockholders may from time to time specify particular provisions of these bylaws, which must not be amended by our board of directors. Our current bylaws were adopted by our board of directors. Therefore, our board of directors can amend our bylaws to make changes to the provisions relating to the quorum requirement and votes requirements to the extent permitted by Delaware Law.
Dividend Rights. The holders of our common stock are entitled to receive such dividends as may be declared by our board of directors out of funds legally available for dividends. Our board of directors is not obligated to declare a dividend. Any future dividends will be subject to the discretion of our board of directors and will depend upon, among other things, future earnings, the operating and financial condition of our company, its capital requirements, general business conditions and other pertinent factors. We do not anticipate that dividends will be paid in the foreseeable future.
Miscellaneous Rights and Provisions. In the event of our liquidation or dissolution, whether voluntary or involuntary, each share of our common stock is entitled to share ratably in any assets available for distribution to holders of our common stock after satisfaction of all liabilities.
Our common stock is not convertible or redeemable and has no preemptive, subscription or conversion rights. There are no conversions, redemption, sinking fund or similar provisions regarding our common stock.
Our common stock, after the fixed consideration thereof has been paid or performed, are not subject to assessment, and the holders of our common stock are not individually liable for the debts and liabilities of our company.
Warrants and Rights
On June 4, 2024, we entered into the Securities Purchase Agreement with certain, pursuant to which we agreed to issue and sell, 715,000 shares of our Common Stock, at a per share price of $1.00 and warrants to purchase up to an additional 1,144,000 shares of Common Stock at a per share exercise price of $1.00. The June 2024 Private Placement closed on June 7, 2024, and we received aggregate gross proceeds of $715,000.
The warrants are exercisable beginning on the six (6) month anniversary of their issuance, have a term of five years from the initial exercise date and entitle the holders to purchase up to 1,144,000 shares of Common Stock. The warrants have an exercise price of $1.00 per share and contain a one-time dilution protection in the event we sell securities at a price less than the then exercise price in effect in a public offering in conjunction with a listing on a national securities exchange.
The securities issued in the June 2024 Private Placement are exempt from the registration requirements of Securities Act pursuant to Section 4(a)(2) of the Securities Act and/or Rule 506(b) of Regulation D promulgated thereunder and pursuant to Regulation S of the Securities Act to non-U.S. investors, because, among other things, the transaction did not involve a public offering, the investors are accredited investors, the investors are taking the securities for investment and not resale and we took appropriate measures to restrict the transfer of the securities. The securities have not been registered under the Securities Act and may not be sold in the United States absent registration or an exemption from registration. The Investors in the June 2024 Private Placement received piggyback registration rights for their Common Stock and the underlying associated Warrants.
| 96 |
The Purchase Agreement contains representations and warranties that the parties made to, and solely for the benefit of, the others in the context of all of the terms and conditions of that agreement and in the context of the specific relationship between the parties.
Description of Securities in this Offering
Public Warrants
Upon completion of this offering we expect to have an additional [_] Warrants outstanding ([_] if the Units reserved for the over-allotment are sold), each Warrant is exercisable for one share of common stock at an exercise price of 100% of the price of each unit sold in the offering and is exercisable at any time up for a period of [five years] following the date of issuance.
The number of Warrants outstanding, and the exercise price of those securities, will be adjusted proportionately in the event of a reverse or forward stock split of our common stock, a recapitalization or reclassification of our common stock, payment of dividends or distributions in common stock to our common stockholders, or similar transactions. In the event that the Company effects a rights offering to its common stock holders or a pro rata distribution of its assets among its common stock holders, then the holder of the Warrants will have the right to participate in such distribution and rights offering to the extent of their pro rata share of the Company’s outstanding common stock assuming they owned the number of shares of common stock issuable upon the exercise of their Warrants. In the event of a “Fundamental Transaction” by the Company, such as a merger or consolidation of it with another company, the sale or other disposition of all or substantially all of the Company’s assets in one or a series of related transactions, a purchase offer, tender offer or exchange offer, or any reclassification, reorganization or recapitalization of the Company’s common stock, then the Warrant holder will have the right to receive, for each share of common stock issuable upon the exercise of the Warrant, at the option of the holder, the number of shares of common stock of the successor or acquiring corporation or of the Company, if it is the surviving corporation, and any additional consideration payable as a result of the Fundamental Transaction, that would have been issued or conveyed to the Warrant holder had the holder exercised the Warrant immediately preceding the closing of the Fundamental Transaction. In lieu of receiving such common stock and additional consideration in the Fundamental Transaction, the Warrant holder may elect to have the Company or the successor entity purchase the Warrant holder’s Warrant for its fair market value measured by the Black Scholes method.
The Company will promptly notify the Warrant holders in writing of any adjustment to the exercise price or to the number of the outstanding Warrants, declaration of a dividend or other distribution, a special non-recurring cash dividend on or a redemption of the common stock, the authorization of a rights offering, the approval of the stock holders required for any proposed reclassification of the common stock, a consolidation or merger by the Company, sale of all or substantially all of the assets of the Company, any compulsory share exchange, or the authorization of any voluntary or involuntary dissolution, liquidation, or winding up of the Company.
The Warrants contain a contractual provision stating that all questions concerning the construction, validity, enforcement and interpretation of the Warrants are governed by and construed and enforced in accordance with the internal laws of the State of New York, without regard to the principles of conflicts of law.
Representative’s Warrants
We also expect to have up to an additional [_] common stock purchase warrants outstanding ([_] if the Units reserved for the over-allotment are sold), issuable to the underwriter of this offering (“Underwriter’s Warrants”). Each Underwriter’s Warrant is exercisable for one share of common stock on a cash or cashless basis at an exercise price of 110% of the price of each unit share of sold in the offering. The Underwriter’s Warrants will be non-exercisable for six (6) months after the effective date (the “Effective Date”) of the registration statement of which this Prospectus forms a part of this offering and will expire five years from the commencement of sales of this offering. The Underwriter’s Warrants will be subject to a lock-up for 360 days from the commencement of sales of this offering including the mandatory lock-up period in accordance with FINRA Rule 5110(e)(1) plus an additional 180-day period. The Underwriter’s Warrants will contain provisions for piggyback registration rights for a period of five years from the commencement of sales of this offering at the Company’s expense.
| 97 |
The number of Underwriter’s Warrants outstanding and the exercise price of those securities will be adjusted proportionately, as permitted by FINRA Rule 5110(g)(8)(E), in the event of a reverse or forward stock split of our common stock, a recapitalization or reclassification of our common stock, payment of dividends or distributions in common stock to our common stockholders, or similar transactions. In the event that the Company effects a rights offering to its common stock holders or a pro rata distribution of its assets among its common stock holders, then the holder of the Underwriter’s Warrants will have the right to participate in such distribution and rights offering to the extent of their pro rata share of the Company’s outstanding common stock assuming they owned the number of shares of common stock issuable upon the exercise of their warrants. In the event of a “Fundamental Transaction” by the Company, such as a merger or consolidation of it with another company, the sale or other disposition of all or substantially all of the Company’s assets in one or a series of related transactions, a purchase offer, tender offer or exchange offer, or any reclassification, reorganization or recapitalization of the Company’s common stock, then the warrant holder will have the right to receive, for each share of common stock issuable upon the exercise of the warrant, at the option of the holder, the number of shares of common stock of the successor or acquiring corporation or of the Company, if it is the surviving corporation, and any additional consideration payable as a result of the Fundamental Transaction that would have been issued or conveyed to the warrant holder had the holder exercised the warrant immediately preceding the closing of the Fundamental Transaction. In lieu of receiving such common stock and additional consideration in the Fundamental Transaction, the warrant holder may elect to have the Company, or the successor entity purchase the warrant holder’s warrant for its fair market value measured by the Black Scholes method.
The Company will promptly notify the holders of the Underwriter’s Warrants in writing of any adjustment to the exercise price or to the number of the outstanding warrants, declaration of a dividend or other distribution, a special non-recurring cash dividend on or redemption of the common stock, the authorization of a rights offering, the approval of the stock holders required for any proposed reclassification of the common stock, a consolidation or merger by the Company, sale of all or substantially all of the assets of the Company, any compulsory share exchange, or the authorization of any voluntary or involuntary dissolution, liquidation, or winding up of the Company.
Transfer Agent and Registrar
The transfer agent and registrar for our common stock is VStock Transfer Company located at 18 Lafayette Pl, Woodmere, NY 11598.
Certain Effects of Authorized but Unissued Stock
We have shares of common stock available for future issuance without stockholder approval. We may issue these additional shares for a variety of corporate purposes, including future public or private offerings to raise additional capital or to facilitate corporate acquisitions or for payment as a dividend on our capital stock.
Anti-Takeover Effects of Provisions of Our Charter Documents
Provisions of our Amended and Restated Articles of Incorporation and Amended and Restated Bylaws may have an anti-takeover effect and may delay, defer or prevent a merger, acquisition, tender offer, takeover attempt or other change of control transaction that a stockholder might consider to be in its interests, including attempts that might result in a premium over the market price for the shares held by our stockholders.
These provisions provide, among other things:
| ● | a classified Board of Directors with staggered three-year terms; | |
| ● | advance notice for nominations of directors by stockholders and for stockholders to include matters to be considered at stockholder meetings; | |
| ● | certain limitations on convening special stockholder meetings and the prohibition of stockholder action by written consent; and | |
| ● | directors may only be removed for cause and only by the affirmative vote of the holders of at least eighty percent (80%) of the voting power of all of the then-outstanding shares of our capital stock entitled to vote at an election of directors, voting together as a single class. |
These anti-takeover provisions, including those noted above, could make it more difficult for a third party to acquire us, even if the third party’s offer may be considered beneficial by many of our stockholders. As a result, our stockholders may be limited in their ability to obtain a premium for their shares.
| 98 |
SHARES ELIGIBLE FOR FUTURE SALE
Prior to this offering, there has been a limited public market for our common stock. Future sales of substantial amounts of common stock in the public market, or the perception that such sales may occur, could adversely affect the market price of our common stock. Although we will apply for listing on Nasdaq, we cannot assure you that our application will be approved or that there will be an active public market for our common stock.
All shares of common stock sold in this offering will be freely tradable without restriction or further registration under the Securities Act, except for any shares of common stock purchased by our “affiliates,” as that term is defined in Rule 144 under the Securities Act or any shares subject to lock-up agreements. Shares purchased by our affiliates would be subject to the Rule 144 resale restrictions described below, other than the holding period requirement.
The remaining shares of common stock outstanding after this offering are “restricted securities,” as that term is defined in Rule 144 under the Securities Act. These restricted securities are eligible for public sale only if they are registered under the Securities Act or if they qualify for an exemption from registration under Rule 144 or Rule 701 under the Securities Act, each of which is summarized below.
We may issue shares of common stock from time to time as consideration for future licensing transactions, investments or other corporate purposes and the number of shares of common stock that we may issue may also be significant. We may also grant registration rights covering those shares of common stock issued in connection with any such transaction. See “Registration Rights” below.
Lock-Up Agreements
In connection with this offering, certain of our stockholders and our directors and executive officers have agreed with the Underwriter that, subject to certain customary exceptions, without the prior written consent of the Underwriter, they will not, for 180 days immediately following the closing of this offering (the “Lock-Up Period”), (a) offer, pledge, announce the intention to sell, sell, contract to sell, sell any option or contract to purchase, purchase any option or contract to sell, grant any option, right or warrant to purchase, make any short sale or otherwise transfer or dispose of, directly or indirectly, our common stock or any securities convertible into, exercisable or exchangeable for or that represent the right to receive our common stock; (b) enter into any swap or other agreement that transfers, in whole or in part, any of the economic consequences of ownership of such securities, whether any such transaction is to be settled by delivery of common stock or other securities, in cash or otherwise; (c) make any demand for or exercise any right with respect to the registration of any shares of our common stock or any security convertible into, exercisable or exchangeable for our common stock; or (d) publicly disclose the intention to do any of the foregoing. The Underwriter may, in its sole discretion, permit any such transactions during the Lock-Up Period in whole or in part and at any time, with or without notice.
Upon the expiration of the Lock-Up Period, substantially all of the shares subject to such lock-up restrictions will become eligible for sale, subject to the limitations discussed above.
Rule 144
Rule 144, as currently in effect, generally provides that, as we have been subject to the public company reporting requirements of Section 13 or Section 15(d) of the Exchange Act for at least 90 days, a stockholder who is not deemed to have been one of our affiliates at any time during the preceding 90 days and who has beneficially owned the shares of our capital stock proposed to be sold for at least six months is entitled to sell such our securities in reliance upon Rule 144 without complying with the volume limitation, manner of sale or notice conditions of Rule 144.
Rule 144 also provides that a stockholder who is deemed to have been one of our affiliates at any time during the preceding 90 days and who has beneficially owned our securities that are proposed to be sold for at least six months is entitled to sell such securities in reliance upon Rule 144 within any three month period beginning 90 days after the date of this prospectus a number of shares that does not exceed the greater of the following:
| ● | 1% of the number of shares of our capital stock then outstanding, which will equal [_] shares immediately after the completion of this offering; or |
| ● | the average weekly trading volume of our Common Stock during the four calendar weeks preceding the filing of a notice on Form 144 with respect to such sale. |
Sales of our securities made in reliance upon Rule 144 by a stockholder who is deemed to have been one of our affiliates at any time during the preceding 90 days are also subject to the current public information, manner of sale and notice conditions of Rule 144.
Rule 701
Rule 701 generally allows a stockholder who purchased shares of our common stock pursuant to a written compensatory plan or contract and who is not deemed to have been one of our affiliates during the immediately preceding 90 days to sell these shares in reliance upon Rule 144, but without being required to comply with the public information, holding period, volume limitation, or notice provisions of Rule 144. Rule 701 also permits our affiliates to sell their Rule 701 shares under Rule 144 without complying with the holding period requirements of Rule 144.
Regulation S
Regulation S under the Securities Act provides that shares owned by any person may be sold without registration in the United States, provided that the sale is affected in an offshore transaction and no directed selling efforts are made in the United States (as these terms are defined in Regulation S), subject to certain other conditions. In general, this means that our shares of common stock may be sold in some other manner outside the United States without requiring registration in the United States.
| 99 |
MATERIAL U.S. FEDERAL INCOME TAX CONSIDERATIONS
The following is a summary of the material U.S. federal income tax consequences of the acquisition, ownership and disposition of the Common Stock (which units or components thereof we sometimes refer to as our “securities” and holders thereof as “holders”), and the acquisition, ownership, exercise, expiration or disposition of the Warrants, but does not purport to be a complete analysis of all the potential tax considerations relating thereto. This summary is based upon the provisions of the Internal Revenue Code of 1986, as amended, or the Code, Treasury Regulations promulgated thereunder, administrative rulings and judicial decisions, all as of the date hereof. These authorities may be changed or subject to differing interpretations, possibly with retroactive effect, so as to result in U.S. federal income tax consequences different from those set forth below. We have not sought and will not seek any ruling from the Internal Revenue Service, or the IRS, with respect to the statements made and the conclusions reached in the following summary, and there can be no assurance that the IRS or a court will agree with such statements and conclusions.
This summary also does not address the tax considerations arising under the laws of any U.S. state or local or any non-U.S. jurisdiction, estate or gift tax, the 3.8% Medicare tax on net investment income or any alternative minimum tax consequences. In addition, this discussion does not address tax considerations applicable to a holder’s particular circumstances or to a holder that may be subject to special tax rules, including, without limitation:
| ● | banks, insurance companies or other financial institutions; |
| ● | tax-exempt or government organizations; |
| ● | brokers or dealers in securities or currencies; |
| ● | traders in securities that elect to use a mark-to-market method of accounting for their securities holdings; |
| ● | persons that own, or are deemed to own, more than five percent of our capital stock; |
| ● | certain U.S. expatriates, citizens or former long-term residents of the United States; |
| ● | persons who hold our shares of Common Stock or Warrants as a position in a hedging transaction, “straddle,” “conversion transaction,” synthetic security, other integrated investment, or other risk reduction transaction; |
| ● | persons who do not hold our Common Stock or Warrants as a capital asset within the meaning of Section 1221 of the Code (generally, for investment purposes); |
| ● | persons deemed to sell our Common Stock or Warrants under the constructive sale provisions of the Code; |
| ● | pension plans; |
| ● | investors in any such entities; |
| ● | persons for whom our stock constitutes “qualified small business stock” within the meaning of Section 1202 of the Code; |
| ● | integral parts or controlled entities of foreign sovereigns; |
| ● | controlled foreign corporations; |
| ● | passive foreign investment companies and corporations that accumulate earnings to avoid U.S. federal income tax; or |
| ● | persons that acquire our Common Stock or Warrants as compensation for services. |
| 100 |
In addition, if a partnership, including any entity or arrangement classified as a partnership for U.S. federal income tax purposes, holds our securities, the tax treatment of a partner generally will depend on the status of the partner, the activities of the partnership, and certain determinations made at the partner level. Accordingly, partnerships that hold our securities, and partners in such partnerships, should consult their tax advisors regarding the U.S. federal income tax consequences to them of the purchase, ownership, and disposition of our securities.
You are urged to consult your tax advisor with respect to the application of the U.S. federal income tax laws to your particular situation, as well as any tax consequences of the purchase, ownership and disposition of our securities arising under the U.S. federal estate or gift tax rules or under the laws of any U.S. state or local or any non-U.S. or other taxing jurisdiction or under any applicable tax treaty.
Definition of a U.S. Holder
For purposes of this summary, a “U.S. Holder” is any beneficial owner of our securities that is a “U.S. person,” and is not a partnership, or an entity treated as a partnership or disregarded from its owner, each for U.S. federal income tax purposes. A U.S. person is any person that, for U.S. federal income tax purposes, is or is treated as any of the following: (a) a citizen or individual resident of the United States, (b) a corporation (or other entity or arrangement treated as a corporation for U.S. federal income tax purposes) created in, or organized under the laws of, the United States or any state thereof or the District of Columbia, (c) an estate whose income is subject to United States federal income tax regardless of its source, or (d) a trust (i) the administration of which is subject to the primary supervision of a U.S. court and which has one or more United States persons (within the meaning of Section 7701(a)(30) of the Code) who have the authority to control all substantial decisions of the trust or (ii) that has otherwise elected to be treated as a United States person under the Code.
For purposes of this summary, a “non-U.S. Holder” is any beneficial owner of our securities that is not a U.S. Holder or a partnership, or other entity treated as a partnership or disregarded from its owner, each for U.S. federal income tax purposes.
Allocation of Purchase Price and Characterization of a Unit
No statutory, administrative or judicial authority directly addresses the treatment of a Unit or instruments similar to a Unit for U.S. federal income tax purposes, and therefore, that treatment is not entirely clear. The acquisition of a Unit should be treated for U.S. federal income tax purposes as the acquisition of one share of our Common Stock or Pre-Funded Warrants, as applicable, and one Warrant. We intend to treat the acquisition of a Unit in this manner and, by purchasing a Unit, you must adopt such treatment for tax purposes. For U.S. federal income tax purposes, each holder of a Unit must allocate the purchase price paid by such holder for such Unit between the share of our Common Stock, as applicable, and the Warrant based on the relative fair market value of each at the time of issuance. The price allocated to each share of our Common Stock and Warrant should be the shareholder’s tax basis in such share of our Common Stock or Warrant. Any disposition of a Unit should be treated for U.S. federal income tax purposes as a disposition of a share of our Common Stock, as applicable, and the Warrant comprising the Unit and, and the amount realized on the disposition should be allocated between the share of Common Stock, as applicable, and the Warrant based on their respective relative fair market values. The separation of a share of our Common Stock and the Warrant constituting a Unit, as applicable, should not be a taxable event for U.S. federal income tax purposes.
The foregoing treatment of the Unit and a holder’s purchase price allocation are not binding on the IRS or the courts. Because there are no authorities that directly address instruments that are similar to the Units, no assurance can be given that the IRS or the courts will agree with the characterization described above or the discussion below. Accordingly, each prospective investor is urged to consult its own tax advisor regarding the tax consequences of an investment in a Unit (including alternative characterizations thereof). The balance of this discussion assumes that the characterization of the Units described above is respected for U.S. federal income tax purposes.
| 101 |
Tax Consequences to U.S. Holders
Distributions on Common Stock
As discussed above under “Dividend Policy,” we do not currently expect to make distributions on our Common Stock. In the event that we do make distributions of cash or other property, distributions paid on Common Stock, other than certain pro rata distributions of Common Stock, will be treated as a dividend to the extent paid out of our current or accumulated earnings and profits and will be includible in income by the U.S. Holder and taxable as ordinary income when received. If a distribution exceeds our current and accumulated earnings and profits, the excess will be first treated as a tax-free return of the U.S. Holder’s investment, up to the U.S. Holder’s tax basis in the Common Stock. Any remaining excess will be treated as a capital gain. Subject to applicable limitations, dividends paid to certain non-corporate U.S. Holders may be eligible for taxation as “qualified dividend income” and therefore may be taxable at rates applicable to long-term capital gains. U.S. Holders should consult their tax advisers regarding the availability of the reduced tax rate on dividends in their particular circumstances. Dividends received by a corporate U.S. Holder will be eligible for the dividends-received deduction if the U.S. Holder meets certain holding period and other applicable requirements.
Constructive Dividends on Warrants
Under Section 305 of the Code, an adjustment to the number of shares of Common Stock that will be issued on the exercise of the Warrants, or an adjustment to the exercise price of the Warrants, may be treated as a constructive distribution to a U.S. Holder of the Warrants if, and to the extent that, such adjustment has the effect of increasing such U.S. Holder’s proportionate interest in our “earnings and profits” or assets, depending on the circumstances of such adjustment (for example, if such adjustment is to compensate for a distribution of cash or other property to our stockholders). Adjustments to the exercise price of a Warrant made pursuant to a bona fide reasonable adjustment formula that has the effect of preventing dilution of the interest of the holders of the Warrants should generally not result in a constructive distribution. Any constructive distributions would generally be subject to the tax treatment described above under “Dividends on Common Stock.”
Sale or Other Disposition of Common Stock
For U.S. federal income tax purposes, gain or loss realized on the sale or other disposition of Common Stock will be capital gain or loss and will be long-term capital gain or loss if the U.S. Holder held the Common Stock for more than one year. The amount of the gain or loss will equal the difference between the U.S. Holder’s tax basis in the Common Stock disposed of and the amount realized on the disposition (or, if the shares of Common Stock, Pre-Funded Warrants or Warrants are held as part of Units, as applicable, at the time of the disposition, the portion of the amount realized on such disposition that is allocated to the shares of Common Stock or Warrants based upon the then fair market values of the shares of Common Stock and Warrants included in the Units, as applicable). Long-term capital gains recognized by non-corporate U.S. Holders will be subject to reduced tax rates. The deductibility of capital losses is subject to limitations.
Sale or Other Disposition, Exercise or Expiration of Warrants
For U.S. federal income tax purposes, gain or loss realized on the sale or other disposition of a Warrant (other than by exercise) will be capital gain or loss and will be long-term capital gain or loss if the U.S. Holder held the Warrant for more than one year at the time of the sale or other disposition. The amount of the gain or loss will equal the difference between the U.S. Holder’s tax basis in the Warrant disposed of and the amount realized on the disposition.
In general, a U.S. Holder will not be required to recognize income, gain or loss upon the exercise of a Warrant by payment of the exercise price, except to the extent of cash paid in lieu of a fractional share. A U.S. Holder’s tax basis in a share of Common Stock received upon exercise will be equal to the sum of (1) the U.S. Holder’s tax basis in the Warrant and (2) the exercise price of the Warrant. A U.S. Holder’s holding period in the stock received upon exercise will commence on the day or the day after such U.S. Holder exercises the Warrant. No discussion is provided herein regarding the U.S. federal income tax treatment on the exercise of a Warrant on a cashless basis, and U.S. Holders are urged to consult their tax advisors as to the exercise of a Warrant on a cashless basis.
| 102 |
If a Warrant expires without being exercised, a U.S. Holder will recognize a capital loss in an amount equal to such U.S. Holder’s tax basis in the Warrant. This loss will be long-term capital loss if, at the time of the expiration, the U.S. Holder’s holding period in the Warrant is more than one year. The deductibility of capital losses is subject to limitations.
FOR NON-U.S. HOLDERS
The following is a general discussion of the material U.S. federal income tax considerations applicable to non-U.S. holders (as defined herein) with respect to their ownership and disposition of our securities issued pursuant to this offering. All prospective non-U.S. holders of our securities should consult their tax advisors with respect to the U.S. federal, state, local and non-U.S. tax consequences of the purchase, ownership and disposition of our securities. In general, a non-U.S. holder means a beneficial owner of our Common Stock (other than a partnership or an entity or arrangement treated as a partnership for U.S. federal income tax purposes) that is not, for U.S. federal income tax purposes:
| ● | an individual who is a citizen or resident of the United States; |
| ● | a corporation, or an entity treated as a corporation for U.S. federal income tax purposes, created or organized in the United States or under the laws of the United States or of any state thereof or the District of Columbia; |
| ● | an estate, the income of which is subject to U.S. federal income tax regardless of its source; or |
| ● | a trust if (1) a U.S. court can exercise primary supervision over the trust’s administration and one or more U.S. persons have the authority to control all of the trust’s substantial decisions or (2) the trust has a valid election in effect under applicable U.S. Treasury Regulations to be treated as a U.S. person. |
| 103 |
This discussion is based on current provisions of the U.S. Internal Revenue Code of 1986, as amended, which we refer to as the Code, existing U.S. Treasury Regulations promulgated thereunder, published administrative pronouncements and rulings of the U.S. Internal Revenue Service, which we refer to as the IRS, and judicial decisions, all as in effect as of the date of this prospectus. These authorities are subject to change and to differing interpretation, possibly with retroactive effect. Any change or differing interpretation could alter the tax consequences to non-U.S. holders described in this prospectus.
We assume in this discussion that a non-U.S. holder holds shares of our securities as a capital asset within the meaning of Section 1221 of the Code (generally, for investment). This discussion does not address all aspects of U.S. federal income taxation that may be relevant to a particular non-U.S. holder in light of that non-U.S. holder’s individual circumstances, nor does it address any alternative minimum, Medicare contribution, estate or gift tax consequences, or any aspects of U.S. state, local or non-U.S. taxes. This discussion also does not consider any specific facts or circumstances that may apply to a non-U.S. holder and does not address the special tax rules applicable to particular non-U.S. holders, such as holders that own, or are deemed to own, more than 5% of our capital stock (except to the extent specifically set forth below), corporations that accumulate earnings to avoid U.S. federal income tax, tax-exempt organizations, banks, financial institutions, insurance companies, brokers, dealers or traders in securities, commodities or currencies, tax-qualified retirement plans, holders who hold or receive our Common Stock pursuant to the exercise of employee stock options or otherwise as compensation, holders holding our Common Stock as part of a hedge, straddle or other risk reduction strategy, conversion transaction or other integrated investment, holders deemed to sell our Common Stock under the constructive sale provisions of the Code, controlled foreign corporations, passive foreign investment companies and certain former U.S. citizens or former long-term residents.
In addition, this discussion does not address the tax treatment of partnerships (or entities or arrangements that are treated as partnerships for U.S. federal income tax purposes) or persons that hold our securities through such partnerships. If a partnership, including any entity or arrangement treated as a partnership for U.S. federal income tax purposes, holds our securities, the U.S. federal income tax treatment of a partner in such partnership will generally depend upon the status of the partner and the activities of the partnership. Such partners and partnerships should consult their tax advisors regarding the tax consequences of the purchase, ownership and disposition of our securities.
There can be no assurance that a court or the IRS will not challenge one or more of the tax consequences described herein, and we have not obtained, nor do we intend to obtain, a ruling with respect to the U.S. federal income tax consequences to a non-U.S. holder of the purchase, ownership or disposition of our securities.
Distributions
As discussed in the section entitled “Dividend Policy,” we do not anticipate paying any dividends on our Common Stock in the foreseeable future. If we make distributions on our Common Stock or on the Warrants (as described above under “Constructive Dividends on Warrants”), those payments will constitute dividends for U.S. federal income tax purposes to the extent we have current or accumulated earnings and profits, as determined under U.S. federal income tax principles. To the extent those distributions exceed both our current and our accumulated earnings and profits, they will constitute a return of capital and will first reduce a Non-U.S. Holder’s basis in our Common Stock or the Warrants, as applicable, but not below zero. Any excess will be treated as capital gain and will be treated as described below under “Gain on Sale or Other Disposition of Common Stock or Warrants.” Any such distributions would be subject to the discussions below regarding back-up withholding and the Foreign Account Tax Compliance Act, or FATCA.
Subject to the discussion below on effectively connected income, any dividend paid to a non-U.S. Holder generally will be subject to U.S. withholding tax either at a rate of 30% of the gross amount of the dividend or such lower rate as may be specified by an applicable income tax treaty. To receive a reduced treaty rate, a non-U.S. Holder must provide us or our agent with an IRS Form W-8BEN, IRS Form W-8 BEN-E or another appropriate version of IRS Form W-8 (or a successor form), which must be updated periodically, and which, in each case, must certify qualification for the reduced rate. Non-U.S. Holders should consult their tax advisors regarding their entitlement to benefits under any applicable income tax treaty.
| 104 |
Dividends paid to a non-U.S. Holder that are effectively connected with the Non-U.S. Holder’s conduct of a trade or business within the United States and that are not eligible for relief from U.S. (net basis) income tax under an applicable income tax treaty generally are exempt from the (gross basis) withholding tax described above. To obtain this exemption from withholding tax, the non-U.S. Holder must provide the applicable withholding agent with an IRS Form W-8ECI or successor form or other applicable IRS Form W-8 certifying that the dividends are effectively connected with the Non-U.S. Holder’s conduct of a trade or business within the United States. Such effectively connected dividends, if not eligible for relief under a tax treaty, would not be subject to a withholding tax, but would be taxed at the same graduated rates applicable to U.S. persons, net of certain deductions and credits and if, in addition, the Non-U.S. Holder is a corporation, may also be subject to a branch profits tax at a rate of 30% (or such lower rate as may be specified by an applicable income tax treaty).
If you are eligible for a reduced rate of withholding tax pursuant to a tax treaty, you may be able to obtain a refund of any excess amounts withheld if you timely file an appropriate claim for refund with the IRS.
Exercise or Expiration of Warrants
In general, a non-U.S. Holder will not be required to recognize income, gain or loss upon the exercise of a Warrant by payment of the exercise price, except possibly to the extent of cash paid in lieu of a fractional share. However, no discussion is provided herein regarding the U.S. federal income tax treatment on the exercise of a Warrant on a cashless basis, and non-U.S. Holders are urged to consult their tax advisors as to the exercise of a Warrant on a cashless basis.
If a Warrant expires without being exercised, a Non-U.S. Holder that is engaged in a U.S. trade or business to which any income from the Warrant would be effectively connected or who is present in the United States for a period or periods aggregating 183 days or more during the calendar year in which the expiration occurs (and certain other conditions are met) will recognize a capital loss in an amount equal to such Non-U.S. Holder’s tax basis in the Warrant. The amount paid to purchase our Common Stock and Warrants will be apportioned between them in proportion to the respective fair market values of the Common Stock and Warrants, and the apportioned amount will be the tax basis of the Common Stock and Warrants respectively. The fair market value of our Common Stock for this purpose will generally be its trading value immediately after issuance.
Gain on Sale, Exchange or Other Disposition of Our Common Stock or Warrants
Subject to the discussion below regarding backup withholding and FATCA, a Non-U.S. Holder generally will not be required to pay U.S. federal income tax on any gain realized upon the sale or other disposition of our Common Stock or the Warrants unless:
| ● | the gain is effectively connected with the Non-U.S. Holder’s conduct of a trade or business within the United States and not eligible for relief under an applicable income tax treaty, in which case the Non-U.S. Holder will be required to pay tax on the net gain derived from the sale under regular graduated U.S. federal income tax rates, and for a Non-U.S. Holder that is a corporation, such Non-U.S. Holder may be subject to the branch profits tax at a 30% rate (or such lower rate as may be specified by an applicable income tax treaty) on such effectively connected gain, as adjusted for certain items; |
| ● | the Non-U.S. Holder is an individual who is present in the United States for a period or periods aggregating 183 days or more during the calendar year in which the sale or disposition occurs and certain other conditions are met, in which case the Non-U.S. Holder will be required to pay a flat 30% tax on the gain derived from the sale, which tax may be offset by U.S. source capital losses (even though the Non-U.S. Holder is not considered a resident of the United States) (subject to applicable income tax or other treaties); or |
| 105 |
| ● | we are a “U.S. real property holding corporation” for U.S. federal income tax purposes, or a USRPHC, at any time within the shorter of the five-year period preceding the disposition or the Non-U.S. Holder’s holding period for our Common Stock or the Warrants. We believe we are not currently and do not anticipate becoming a USRPHC. However, because the determination of whether we are a USRPHC depends on the fair market value of our United States real property interests relative to the fair market value of our other business assets, there can be no assurance that we will not become a USRPHC in the future. Even if we become a USRPHC, however, gain arising from the sale or other taxable disposition by a Non-U.S. Holder of our Common Stock will not be subject to United States federal income tax if (A) in the case of our Common Stock, (a) shares of our Common Stock are “regularly traded,” as defined by applicable Treasury Regulations, on an established securities market, such as Nasdaq, and (b) the Non-U.S. Holder owns or owned, actually and constructively, 5% or less of the shares of our Common Stock throughout the five-year period ending on the date of the sale or exchange; and (B) in the case of the Warrants, either (a)(i) shares of our Common Stock are “regularly traded,” as defined by applicable Treasury Regulations, on an established securities market, such as Nasdaq, (ii) the Warrants are not considered regularly traded on an established securities market and (iii) the Non-U.S. Holder does not own, actually or constructively, Warrants with a fair market value greater than the fair market value of 5% of the shares of our Common Stock, determined as of the date that such Non-U.S. Holder acquired its Warrants, or (b)(i) the Warrants are considered regularly traded on an established securities market, and (ii) the Non-U.S. Holder owns or owned, actually and constructively, 5% or less of the Warrants throughout the five-year period ending on the date of the sale or exchange. The Warrants are not expected to be regularly traded on an established securities market. If the foregoing exception does not apply, and we are a USRPHC, such Non-U.S. Holder’s proceeds received on the disposition of shares will generally be subject to withholding at a rate of 15% and such Non-U.S. Holder will generally be taxed on any gain in the same manner as gain that is effectively connected with the conduct of a U.S. trade or business, except that the branch profits tax generally will not apply. |
Backup Withholding and Information Reporting
Information returns may be filed with the IRS in connection with distributions on our Common Stock or constructive dividends on the Warrants, and the proceeds of a sale or other disposition of the Common Stock or the Warrants. A non-exempt U.S. Holder may be subject to U.S. backup withholding on these payments if it fails to provide its taxpayer identification number to the withholding agent and comply with certification procedures or otherwise establish an exemption from backup withholding.
A Non-U.S. Holder may be subject to U.S. information reporting and backup withholding on these payments unless the non-U.S. Holder complies with certification procedures to establish that it is not a U.S. person (within the meaning of the Code). The certification requirements generally will be satisfied if the non-U.S. Holder provides the applicable withholding agent with a statement on the applicable IRS Form W-8BEN or IRS Form W-8BEN-E (or suitable substitute or successor form), together with all appropriate attachments, signed under penalties of perjury, stating, among other things, that such non-U.S. Holder is not a U.S. Person. Applicable Treasury Regulations provide alternative methods for satisfying this requirement. In addition, the amount of distributions on common stock or constructive dividends on common stock paid to a non-U.S. Holder, and the amount of any U.S. federal tax withheld therefrom, must be reported annually to the IRS and the holder. This information may be made available by the IRS under the provisions of an applicable tax treaty or agreement to the tax authorities of the country in which the non-U.S. Holder resides.
Payment of the proceeds of the sale or other disposition of the Common Stock or the Warrants to or through a non-U.S. office of a U.S. broker or of a non-U.S. broker with certain specified U.S. connections generally will be subject to information reporting requirements, but not backup withholding, unless the non-U.S. Holder certifies under penalties of perjury that it is not a U.S. person, or an exemption otherwise applies. Payments of the proceeds of a sale or other disposition of the Common Stock or the Warrants to or through a U.S. office of a broker generally will be subject to information reporting and backup withholding, unless the non-U.S. Holder certifies under penalties of perjury that it is not a U.S. person or otherwise establishes an exemption.
Backup withholding is not an additional tax. The amount of any backup withholding from a payment generally will be allowed as a credit against the holder’s U.S. federal income tax liability and may entitle the holder to a refund, provided that the required information is timely furnished to the IRS.
| 106 |
Foreign Account Tax Compliance Act
FATCA imposes withholding tax on certain types of payments made to foreign financial institutions and certain other non-U.S. entities. The legislation imposes a 30% withholding tax on dividends on, or, subject to the discussion of certain proposed Treasury Regulations below, gross proceeds from the sale or other disposition of, our Common Stock or the Warrants paid to a “foreign financial institution” or to certain “non-financial foreign entities” (each as defined in the Code), unless (i) the foreign financial institution undertakes certain diligence and reporting obligations, (ii) the non-financial foreign entity either certifies it does not have any “substantial United States owners” (as defined in the Code) or furnishes identifying information regarding each substantial United States owner, or (iii) the foreign financial institution or non-financial foreign entity otherwise qualifies for an exemption from these rules. If the payee is a foreign financial institution and is subject to the diligence and reporting requirements in (i) above, it must enter into an agreement with the U.S. Treasury requiring, among other things, that it undertake to identify accounts held by “specified United States persons” or “United States-owned foreign entities” (each as defined in the Code), annually report certain information about such accounts, and withhold 30% on payments to account holders whose actions prevent it from complying with these reporting and other requirements. If the country in which a payee is resident has entered into an “intergovernmental agreement” with the United States regarding FATCA, that agreement may permit the payee to report to that country rather than to the U.S. Department of the Treasury. The U.S. Treasury recently released proposed Treasury Regulations which, if finalized in their present form, would eliminate the federal withholding tax of 30% applicable to the gross proceeds of a sale or other disposition of our Common Stock or the Warrants. In its preamble to such proposed Treasury Regulations, the U.S. Treasury stated that taxpayers may generally rely on the proposed regulations until final regulations are issued. Prospective investors should consult their own tax advisors regarding the possible impact of these rules on their investment in our Common Stock or the Warrants, and the possible impact of these rules on the entities through which they hold our Common Stock or the Warrants, including, without limitation, the process and deadlines for meeting the applicable requirements to prevent the imposition of this 30% withholding tax under FATCA.
THE PRECEDING DISCUSSION IS FOR GENERAL INFORMATION ONLY. EACH PROSPECTIVE INVESTOR SHOULD CONSULT ITS TAX ADVISOR REGARDING THE PARTICULAR U.S. FEDERAL, STATE AND LOCAL AND NON-U.S. TAX CONSEQUENCES OF PURCHASING, HOLDING AND DISPOSING OF OUR COMMON STOCK AND WARRANTS, INCLUDING THE CONSEQUENCES OF ANY PROPOSED CHANGE IN APPLICABLE LAWS.
| 107 |
UNDERWRITING
We will enter into an underwriting agreement with Maxim Group LLC as the sole representative of the underwriters (“Maxim” or the “Representative”), with respect to the shares and warrants being offered. Maxim is the sole book running manager for the offering. Subject to the terms and conditions of an underwriting agreement between us and the Representative, we will agree to sell to each underwriter named below, and each underwriter named below has severally agreed to purchase, at the public offering price less the underwriting discounts set forth on the cover page of this prospectus, the number of shares of common stock and warrants listed next to its name in the following table:
| Name of Underwriter | Number of Shares | Number of Warrants | ||||||
| Maxim Group LLC | ||||||||
| Total | ||||||||
The underwriters are committed to purchase all the shares of common stock and warrants offered by this prospectus if they purchase any shares of common stock and warrants. The underwriting agreement also provides that if an underwriter defaults, the purchase commitments of non-defaulting underwriters may be increased, or the offering may be terminated. The underwriters are not obligated to purchase the shares of common stock and/or warrants covered by the underwriters’ over-allotment option described below. The underwriters are offering the shares of common stock and warrants, subject to prior sale, when, as and if issued to and accepted by them, subject to approval of legal matters by their counsel, and other conditions contained in the underwriting agreement, such as the receipt by the underwriters of officer’s certificates and legal opinions. The underwriters reserve the right to withdraw, cancel or modify offers to the public and to reject orders in whole or in part.
Over-Allotment Option
We have granted to the underwriters an option, exercisable no later than 45 calendar days after the date of the underwriting agreement, to purchase up to [_] (or 15%) shares of common stock and/or warrants at the public offering price listed on the cover page of this prospectus, less underwriting discounts and commissions. The underwriters may exercise this option only to cover over-allotments, if any, made in connection with this offering. To the extent the option is exercised, and the conditions of the underwriting agreement are satisfied, we will be obligated to sell to the underwriters, and the underwriters will be obligated to purchase, these additional shares of common stock and/or warrants.
Representative’s Warrants
We have agreed to grant to Maxim Group LLC, Underwriter’s Warrants to purchase a number of shares equal to six percent (6.0%) of the total number of shares of common stock sold in this offering, at an exercise price equal to 110% of the price per unit sold in this offering. The Underwriter’s Warrants will contain a cashless exercise feature. Each Underwriter’s Warrant is exercisable for one share of common stock on a cash or cashless basis at an exercise price of 110% of the price of each unit sold in the offering. The Underwriter’s Warrants will be subject to a lock-up for 180 days from the commencement of sales of this offering including the mandatory lock-up period in accordance with FINRA Rule 5110(e) and will be non-exercisable for six (6) months after the Effective Date of the registration statement of which this Prospectus forms a part of this offering, and will expire five (5) years from the commencement of sales of this offering. The Underwriter’s Warrants will contain provisions for piggyback registration rights for a period of five (5) years from the commencement of sales of this offering at the Company’s expense.
The number of Underwriter’s Warrants outstanding, and the exercise price of those securities, will be adjusted proportionately, as permitted by FINRA Rule 5110(g)(8)(E).
| 108 |
Discounts and Commissions; Expenses
The following table shows the public offering price, underwriting discount and proceeds, before expenses, to us. The information assumes either no exercise or full exercise by the Representative of the over-allotment option.
| Per Share(1) | Total Without Over- Allotment Option | Total With Full Over- Allotment Option | ||||||||||
| Public offering price | $ | $ | $ | |||||||||
| Underwriting discount (8%) | $ | $ | $ | |||||||||
| Proceeds, before expenses, to us | $ | $ | $ | |||||||||
| (1) | The fees shown do not include the warrant to purchase shares of common stock issuable to the underwriters at closing. |
The underwriters propose to offer the shares offered by us to the public at the public offering price per share set forth on the cover of this prospectus. In addition, the underwriters may offer some of the shares to other securities dealers at such price less a concession of $ per share. After the initial offering, the public offering price and concession to dealers may be changed.
We paid an expense deposit of $25,000 to the Representative, which will be applied against the accountable expenses that will be paid by us to the Representative in connection with this offering.
We have also agreed to reimburse the Representative for reasonable out-of-pocket expenses not to exceed $150,000. We estimate that total expenses payable by us in connection with this offering, other than the underwriting discount, will be approximately $[_].
Lock-Up Agreements
We and each of our officers, directors, affiliates and certain existing stockholders aggregating at least 5.0% of our outstanding shares have agreed, subject to certain exceptions, not to offer, issue, sell, contract to sell, encumber, grant any option for the sale of or otherwise dispose of any shares of our common stock or other securities convertible into or exercisable or exchangeable for shares of our common stock for a period of six (6) months after this offering is completed without the prior written consent of Maxim.
Maxim may in its sole discretion and at any time without notice release some or all of the shares subject to lock-up agreements prior to the expiration of the lock-up period. When determining whether or not to release shares from the lock-up agreements, the Representative will consider, among other factors, the security holder’s reasons for requesting the release, the number of shares for which the release is being requested and market conditions at the time.
Right of First Refusal
We have granted Maxim a right of first refusal, for a period of eighteen (18) months from the commencement of sales of this offering, to act as sole managing underwriter and sole book-runner, sole placement agent or sole sales agent, at Maxim’s sole and exclusive discretion, for any and all future public or private equity, equity-linked or debt (excluding commercial bank debt) offerings for which the Company retains the service of an underwriter, agent, advisor, finder or other person or entity in connection with such offering during such (each, a “Subject Transaction”), during such eighteen (18) month period, of the Company, or any successor to or subsidiary of the Company, on terms and conditions customary to the Maxim for such Subject Transactions.
Indemnification
We have agreed to indemnify the underwriters against certain liabilities, including liabilities under the Securities Act, and to contribute to payments that the underwriters may be required to make for these liabilities.
| 109 |
OTCQB and Nasdaq Capital Market
Our common stock is presently quoted on the OTCQB marketplace under the symbol “IRME”. We intend to apply to list our common stock and warrants on the Nasdaq under the symbols “IRME.” and “IRMEW”, respectively. No assurance can be given that our application will be approved. Trading Quotes of securities on an over-the-counter marketplace may not be indicative of the market price of those securities on a national securities exchange. There is no established public trading market for the warrants. No assurance can be given that a trading market will develop for the warrants.
Price Stabilization, Short Positions, and Penalty Bids
In connection with this offering, the underwriters may engage in transactions that stabilize, maintain or otherwise affect the price of our common stock. Specifically, the underwriters may over-allot in connection with this offering by selling more shares and warrants than are set forth on the cover page of this prospectus. This creates a short position in our common stock for its own account. The short position may be either a covered short position or a naked short position. In a covered short position, the number of shares common stock or warrants over-allotted by the underwriters is not greater than the number of shares of common stock or warrants that they may purchase in the over-allotment option. In a naked short position, the number of shares of common stock or warrants involved is greater than the number of shares common stock or warrants in the over-allotment option. To close out a short position, the underwriters may elect to exercise all or part of the over-allotment option. The underwriters may also elect to stabilize the price of our common stock or reduce any short position by bidding for, and purchasing, common stock in the open market. Since the warrants will not be listed and are not expected to trade, the underwriters cannot purchase the warrants in the open market and, as a result, the underwriters cannot and will not enter into naked short positions.
The underwriters may also impose a penalty bid. This occurs when a particular underwriter or dealer repays selling concessions allowed to it for distributing a security in this offering because the underwriter repurchases that security in stabilizing or short covering transactions.
Finally, the underwriters may bid for, and purchase, shares of our common stock in market making transactions, including “passive” market making transactions as described below.
These activities may stabilize or maintain the market price of our common stock at a price that is higher than the price that might otherwise exist in the absence of these activities. The underwriters are not required to engage in these activities, and may discontinue any of these activities at any time without notice. These transactions may be effected on Nasdaq, in the over-the-counter market, or otherwise.
In connection with this offering, the underwriters and selling group members, if any, or their affiliates may engage in passive market making transactions in our common stock immediately prior to the commencement of sales in this offering, in accordance with Rule 103 of Regulation M under the Exchange Act. Rule 103 generally provides that:
| ● | a passive market maker may not effect transactions or display bids for our common stock in excess of the highest independent bid price by persons who are not passive market makers; |
| ● | net purchases by a passive market maker on each day are generally limited to 30% of the passive market maker’s average daily trading volume in our common stock during a specified two-month prior period or 200 shares, whichever is greater, and must be discontinued when that limit is reached; and |
| ● | passive market making bids must be identified as such. |
Electronic Distribution
A prospectus in electronic format may be made available on a website maintained by the representatives of the underwriters and may also be made available on a website maintained by other underwriters. The underwriters may agree to allocate a number of shares to underwriters for sale to their online brokerage account holders. Internet distributions will be allocated by the representatives of the underwriters to underwriters that may make Internet distributions on the same basis as other allocations. In connection with the offering, the underwriters or syndicate members may distribute prospectuses electronically. No forms of electronic prospectus other than prospectuses that are printable as Adobe® PDF will be used in connection with this offering.
| 110 |
The underwriters have informed us that they do not expect to confirm sales of shares and warrants offered by this prospectus to accounts over which they exercise discretionary authority.
Other than the prospectus in electronic format, the information on any underwriter’s website and any information contained in any other website maintained by an underwriter is not part of the prospectus or the registration statement of which this prospectus forms a part, has not been approved and/or endorsed by us or any underwriter in its capacity as underwriter and should not be relied upon by investors.
Certain Relationships
Certain of the underwriters and their affiliates may provide, from time to time, investment banking and financial advisory services to us in the ordinary course of business, for which they may receive customary fees and commissions.
Selling Restrictions
Canada. The securities may be sold in Canada only to purchasers purchasing, or deemed to be purchasing, as principal that are accredited investors, as defined in National Instrument 45-106 Prospectus Exemptions or subsection 73.3(1) of the Securities Act (Ontario), and are permitted clients, as defined in National Instrument 31-103 Registration Requirements, Exemptions and Ongoing Registrant Obligations. Any resale of the securities must be made in accordance with an exemption from, or in a transaction not subject to, the prospectus requirements of applicable securities laws.
Securities legislation in certain provinces or territories of Canada may provide a purchaser with remedies for rescission or damages if this prospectus supplement (including any amendment thereto) contains a misrepresentation, provided that the remedies for rescission or damages are exercised by the purchaser within the time limit prescribed by the securities legislation of the purchaser’s province or territory. The purchaser should refer to any applicable provisions of the securities legislation of the purchaser’s province or territory for particulars of these rights or consult with a legal advisor.
Pursuant to section 3A.3 of National Instrument 33-105 Underwriting Conflicts (NI 33-105), the underwriters are not required to comply with the disclosure requirements of NI 33-105 regarding underwriters conflicts of interest in connection with this offering.
European Economic Area. In relation to each Member State of the European Economic Area which has implemented the Prospectus Directive (each, a “Relevant Member State”) an offer to the public of any securities may not be made in that Relevant Member State, except that an offer to the public in that Relevant Member State of any securities may be made at any time under the following exemptions under the Prospectus Directive, if they have been implemented in that Relevant Member State:
| ● | to any legal entity which is a qualified investor as defined in the Prospectus Directive; |
| ● | to fewer than 100 or, if the Relevant Member State has implemented the relevant provision of the 2010 PD Amending Directive, 150, natural or legal persons (other than qualified investors as defined in the Prospectus Directive), as permitted under the Prospectus Directive, subject to obtaining the prior consent of the representatives for any such offer; or |
| ● | in any other circumstances falling within Article 3(2) of the Prospectus Directive, provided that no such offer of securities shall result in a requirement for the publication by us or any underwriters of a prospectus pursuant to Article 3 of the Prospectus Directive. |
For the purposes of this provision, the expression an “offer to the public” in relation to any securities in any Relevant Member State means the communication in any form and by any means of sufficient information on the terms of the offer and any securities to be offered so as to enable an investor to decide to purchase any securities, as the same may be varied in that Member State by any measure implementing the Prospectus Directive in that Member State, the expression “Prospectus Directive” means Directive 2003/71/EC (and amendments thereto, including the 2010 PD Amending Directive, to the extent implemented in the Relevant Member State), and includes any relevant implementing measure in the Relevant Member State, and the expression “2010 PD Amending Directive” means Directive 2010/73/EU.
| 111 |
Israel. This document does not constitute a prospectus under the Israeli Securities Law, 5728-1968, or the Securities Law, and has not been filed with or approved by the Israel Securities Authority. In the State of Israel, this document is being distributed only to, and is directed only at, and any offer of the shares is directed only at, investors listed in the first addendum, or the Addendum, to the Israeli Securities Law, consisting primarily of joint investment in trust funds, provident funds, insurance companies, banks, portfolio managers, investment advisors, members of the Tel Aviv Stock Exchange, underwriters, venture capital funds, entities with equity in excess of NIS 50 million and “qualified individuals”, each as defined in the Addendum (as it may be amended from time to time), collectively referred to as qualified investors (in each case purchasing for their own account or, where permitted under the Addendum, for the accounts of their clients who are investors listed in the Addendum). Qualified investors will be required to submit written confirmation that they fall within the scope of the Addendum, are aware of the meaning of same and agree to it.
United Kingdom. Each underwriter has represented and agreed that:
| ● | it has only communicated or caused to be communicated and will only communicate or cause to be communicated an invitation or inducement to engage in investment activity (within the meaning of Section 21 of the Financial Services and Markets Act 2000 (the FSMA) received by it in connection with the issue or sale of the securities in circumstances in which Section 21(1) of the FSMA does not apply to us; and |
| ● | it has complied and will comply with all applicable provisions of the FSMA with respect to anything done by it in relation to the securities in, from or otherwise involving the United Kingdom. |
Switzerland. The securities may not be publicly offered in Switzerland and will not be listed on the SIX Swiss Exchange (the SIX) or on any other stock exchange or regulated trading facility in Switzerland. This document has been prepared without regard to the disclosure standards for issuance prospectuses under art. 652a or art. 1156 of the Swiss Code of Obligations or the disclosure standards for listing prospectuses under art. 27 ff. of the SIX Listing Rules or the listing rules of any other stock exchange or regulated trading facility in Switzerland. Neither this document nor any other offering or marketing material relating to the securities or the offering may be publicly distributed or otherwise made publicly available in Switzerland.
Neither this document nor any other offering or marketing material relating to the offering, or the securities have been or will be filed with or approved by any Swiss regulatory authority. In particular, this document will not be filed with, and the offer of securities will not be supervised by, the Swiss Financial Market Supervisory Authority FINMA, and the offer of securities has not been and will not be authorized under the Swiss Federal Act on Collective Investment Schemes (CISA). Accordingly, no public distribution, offering or advertising, as defined in CISA, its implementing ordinances and notices, and no distribution to any non-qualified investor, as defined in CISA, its implementing ordinances and notices, shall be undertaken in or from Switzerland, and the investor protection afforded to acquirers of interests in collective investment schemes under CISA does not extend to acquirers of securities.
Australia. No placement document, prospectus, product disclosure statement or other disclosure document has been lodged with the Australian Securities and Investments Commission (ASIC), in relation to the offering.
This prospectus does not constitute a prospectus, product disclosure statement or other disclosure document under the Corporations Act 2001 (the Corporations Act) and does not purport to include the information required for a prospectus, product disclosure statement or other disclosure document under the Corporations Act.
Any offer in Australia of the securities may only be made to persons (the Exempt Investors) who are “sophisticated investors” (within the meaning of section 708(8) of the Corporations Act), “professional investors” (within the meaning of section 708(11) of the Corporations Act) or otherwise pursuant to one or more exemptions contained in section 708 of the Corporations Act so that it is lawful to offer the securities without disclosure to investors under Chapter 6D of the Corporations Act.
| 112 |
The securities applied for by Exempt Investors in Australia must not be offered for sale in Australia in the period of 12 months after the date of allotment under the offering, except in circumstances where disclosure to investors under Chapter 6D of the Corporations Act would not be required pursuant to an exemption under section 708 of the Corporations Act or otherwise or where the offer is pursuant to a disclosure document which complies with Chapter 6D of the Corporations Act. Any person acquiring securities must observe such Australian on-sale restrictions.
This prospectus contains general information only and does not take account of the investment objectives, financial situation or particular needs of any particular person. It does not contain any securities recommendations or financial product advice. Before making an investment decision, investors need to consider whether the information in this prospectus is appropriate to their needs, objectives and circumstances, and, if necessary, seek expert advice on those matters.
Notice to Prospective Investors in the Cayman Islands. No invitation, whether directly or indirectly, may be made to the public in the Cayman Islands to subscribe for our securities.
Taiwan. The securities have not been and will not be registered with the Financial Supervisory Commission of Taiwan pursuant to relevant securities laws and regulations and may not be sold, issued or offered within Taiwan through a public offering or in circumstances which constitutes an offer within the meaning of the Securities and Exchange Act of Taiwan that requires a registration or approval of the Financial Supervisory Commission of Taiwan. No person or entity in Taiwan has been authorized to offer, sell, give advice regarding or otherwise intermediate the offering and sale of the securities in Taiwan.
Notice to Prospective Investors in Hong Kong. The contents of this prospectus have not been reviewed by any regulatory authority in Hong Kong. You are advised to exercise caution in relation to the offer. If you are in any doubt about any of the contents of this prospectus, you should obtain independent professional advice. Please note that (i) our shares may not be offered or sold in Hong Kong, by means of this prospectus or any document other than to “professional investors” within the meaning of Part I of Schedule 1 of the Securities and Futures Ordinance (Cap.571, Laws of Hong Kong) (SFO) and any rules made thereunder, or in other circumstances which do not result in the document being a “prospectus” within the meaning of the Companies Ordinance (Cap.32, Laws of Hong Kong) (CO) or which do not constitute an offer or invitation to the public for the purpose of the CO or the SFO, and (ii) no advertisement, invitation or document relating to our shares may be issued or may be in the possession of any person for the purpose of issue (in each case whether in Hong Kong or elsewhere) which is directed at, or the contents of which are likely to be accessed or read by, the public in Hong Kong (except if permitted to do so under the securities laws of Hong Kong) other than with respect to the shares which are or are intended to be disposed of only to persons outside Hong Kong or only to “professional investors” within the meaning of the SFO and any rules made thereunder.
Notice to Prospective Investors in the People’s Republic of China. This prospectus may not be circulated or distributed in the PRC and the shares may not be offered or sold, and will not offer or sell to any person for re-offering or resale directly or indirectly to any resident of the PRC except pursuant to applicable laws, rules and regulations of the PRC. For the purpose of this paragraph only, the PRC does not include Taiwan and the special administrative regions of Hong Kong and Macau.
Transfer Agent and Registrar
The transfer agent and registrar for our common stock and warrant agent is VStock Transfer LLC, at 18 Lafayette Place, Woodmere, NY 11598. The transfer agent’s telephone number is (212) 828-8436.
| 113 |
LEGAL MATTERS
The validity of the shares of common stock offered in this prospectus is being passed upon for us by Sullivan and Worcester LLP, New York, New York. The Underwriter has been represented in connection with this offering by Ellenoff Grossman & Schole, LLP, New York, New York.
| 114 |
EXPERTS
The consolidated financial statements of IR-Med Inc. as of December 31, 2023 and 2022 and for each of the years in the two-year period ended December 31, 2023, have been included herein in reliance upon the report of Somekh Chaikin, a member firm of KPMG International, independent registered public accounting firm, appearing elsewhere herein, and upon the authority of said firm as experts in accounting and auditing.
The audit report covering the December 31, 2023 consolidated financial statements contains an explanatory paragraph that states that the Company’s recurring losses from operations and accumulated deficit raise substantial doubt about the entity’s ability to continue as a going concern. The consolidated financial statements do not include any adjustments that might result from the outcome of that uncertainty.
| 115 |
WHERE YOU CAN FIND ADDITIONAL INFORMATION
We are subject to the information requirements of the Exchange Act and we therefore file periodic reports, proxy statements and other information with the SEC relating to our business, financial statements and other matters. The reports, proxy statements and other information we file may be inspected and copied at prescribed rates at the SEC’s Public Reference Room located at 100 F Street, N.E., Washington, D.C. 20549. You may obtain information on the operation of the SEC’s Public Reference Room by calling the SEC at 1-800-SEC-0330. The SEC also maintains a website that contains reports, proxy and information statements and other information regarding issuers like us that file electronically with the SEC. The address of the SEC’s website is http://www.sec.gov.
This prospectus constitutes part of a registration statement on Form S-1 filed under the Securities Act with respect to the shares of common stock covered hereby. As permitted by the SEC’s rules, this prospectus omits some of the information, exhibits and undertakings included in the registration statement. You may read and copy the information omitted from this prospectus but contained in the registration statement, as well as the periodic reports and other information we file with the SEC, at the public reference room and web site of the SEC referred to above. You may also access our filings with the SEC on our web site is located at https://www.ir-medical.com/. The information contained on our web site is not part of this prospectus.
| 116 |
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
IR-Med, Inc.
| F-1 |
IR-Med, INC.
and subsidiary
Consolidated Financial Statements
December 31, 2023
| F-2 |
IR-Med, Inc.
Consolidated Financial Statements as of December 31, 2023
| F-3 |

Somekh Chaikin
KPMG Millennium Tower
17 Ha’arba’a Street, PO Box 609
Tel Aviv 61006, Israel
+972 3 684 8000
Report of Independent Registered Public Accounting Firm
To the Stockholders and the Board of Directors of IR Med, Inc.
Opinion on the Consolidated Financial Statements
We have audited the accompanying consolidated balance sheets of IR-Med, Inc. and subsidiary (the Company) as of December 31, 2023 and 2022, the related consolidated statements of operations, changes in stockholders’ equity, and cash flows for each of the years in the two-year period ended December 31, 2023, and the related notes (collectively, the consolidated financial statements). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2023, and 2022, and the results of its operations and its cash flows for each of the years in the two-year period ended December 31, 2023, in conformity with U.S. generally accepted accounting principles.
Going Concern
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 1b to the consolidated financial statements, the Company has suffered recurring losses from operations and has an accumulated deficit that raise substantial doubt about its ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 1b. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these consolidated financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
Somekh Chaikin
Member Firm of KPMG International
We have served as the Company’s auditor since 2020.
Tel Aviv, Israel
March 29, 2024
KPMG Somekh Chaikin, an Israeli partnership and a member firm of the KPMG global organization of independent member firms affiliated with KPMG International Limited, a private English company limited by guarantee.
| F-4 |
IR-Med, Inc.
Consolidated Balance Sheets as of December 31
| 2023 | 2022 | ||||||||||
| Note | US Dollars (In thousands) | ||||||||||
| Assets | |||||||||||
| Current assets | |||||||||||
| Cash and cash equivalents | 3 | ||||||||||
| Accounts receivable | 4 | ||||||||||
| Total current assets | |||||||||||
| Non-current assets | |||||||||||
| Long term restricted deposits | 10 | ||||||||||
| Right of use assets | 6 | ||||||||||
| Property and equipment, net | 5 | ||||||||||
| Total non-current assets | |||||||||||
| Total assets | |||||||||||
| Liabilities and stockholders’ equity | |||||||||||
| Current liabilities | |||||||||||
| Trade and other payables | 7 | ||||||||||
| Stockholders’ loans | 8 | ||||||||||
| Total current liabilities | |||||||||||
| Non-current liabilities | |||||||||||
Stockholders’ loans | 8 | ||||||||||
| Long term lease liability | 6 | ||||||||||
| Total non-current liabilities | |||||||||||
| Total liabilities | |||||||||||
| Contingent liabilities and commitments | 10 | ||||||||||
| Stockholders’ equity | 9 | ||||||||||
| Common stock, par value $ per share, shares authorized: and issued and outstanding as of December 31, 2023, and 2022, respectively | |||||||||||
| Additional paid-in capital | |||||||||||
| Accumulated deficit | ( | ) | ( | ) | |||||||
| Total stockholders’ equity | |||||||||||
| Total liabilities and stockholders’ equity | |||||||||||
The accompanying notes are an integral part of the consolidated financial statements.
| F-5 |
IR-Med, Inc.
Consolidated Statements of Operations
| For the year | For the year | ||||||||||
| ended | ended | ||||||||||
| December 31, | December 31, | ||||||||||
| 2023 | 2022 | ||||||||||
| Note | US Dollars (In thousands) | ||||||||||
| Research and development expenses | 11 | ||||||||||
| Marketing expenses | 12 | ||||||||||
| General and administrative expenses | 13 | ||||||||||
| Total operating loss | |||||||||||
| Financial income, net | 14 | ( | ) | ( | ) | ||||||
| Loss for the year | |||||||||||
| Loss per share | 15 | ||||||||||
| Basic and dilutive loss per common stock (in U.S. dollars) | ) | ) | |||||||||
The accompanying notes are an integral part of the consolidated financial statements.
| F-6 |
IR-Med, Inc.
Consolidated Statement of Changes in Stockholders’ Equity
| Additional | ||||||||||||||||||||
| paid-in | Accumulated | |||||||||||||||||||
| Common Stock | Capital | deficit | Total | |||||||||||||||||
| Number of | ||||||||||||||||||||
| Shares | US dollars (In Thousand) | |||||||||||||||||||
| Balance as of January 1, 2023 | ( | ) | ||||||||||||||||||
| Stock-based compensation | * | |||||||||||||||||||
| Private placement of common stock and warrants | ||||||||||||||||||||
| Loss for the year | ( | ) | ( | ) | ||||||||||||||||
| Balance as of December 31, 2023 | ( | ) | ||||||||||||||||||
| Balance as of January 1, 2022 | ( | ) | ||||||||||||||||||
| Stock-based compensation | * | |||||||||||||||||||
| Private placement of common stock and warrants | ||||||||||||||||||||
| Loss for the year | ( | ) | ( | ) | ||||||||||||||||
| Balance as of December 31, 2022 | ( | ) | ||||||||||||||||||
| (*) |
The accompanying notes are an integral part of the consolidated financial statements.
| F-7 |
IR-Med, Inc.
Consolidated Statements of Cash Flows
| For the year | For the year | |||||||
| ended | ended | |||||||
| December 31, | December 31, | |||||||
| 2023 | 2022 | |||||||
| US Dollars (In thousands) | ||||||||
| Cash flows from operating activities | ||||||||
| Loss for the year | ( | ) | ( | ) | ||||
| Adjustments to reconcile loss for the year to net cash used in operating activities: | ||||||||
| Stock based compensation | ||||||||
| Depreciation | ||||||||
| Accrued financial income | ( | ) | ( | ) | ||||
| Decrease (increase) in accounts receivable | ( | ) | ||||||
| Increase in trade and other payables | ||||||||
| Net cash used in operating activities | ( | ) | ( | ) | ||||
| Cash flows from investing activities | ||||||||
| Purchase of property and equipment | ( | ) | ( | ) | ||||
| Increase in long term deposit | ( | ) | ||||||
| Net cash used in investing activities | ( | ) | ( | ) | ||||
| Cash flows from financing activities | ||||||||
| Proceeds from private placement of common stock and warrants. | ||||||||
| Net cash provided by financing activities | ||||||||
| Effect of exchange rate changes on cash | ( | ) | ( | ) | ||||
| Net (decrease) increase in cash and cash equivalents | ( | ) | ||||||
| Cash and cash equivalents as at the beginning of the year | ||||||||
| Cash and cash equivalents as at the end of the year | ||||||||
| Non-cash financing Activities: | ||||||||
| Increase in right of use assets against lease liability | ||||||||
| Increase in right of use assets against long term restricted deposits | ||||||||
The accompanying notes are an integral part of the consolidated financial statements.
| F-8 |
IR-Med, Inc.
Notes to the Consolidated Financial Statements
Note 1 - General
| A. | Description of Business |
| IR-Med, Inc. (OTC QB: IRME, hereinafter: the “Parent Company”) was incorporated in Nevada in 2007. IR-Med, Inc. was previously named International Display Advertising, Inc. and changed its name to IR-Med, Inc. in January 2021. | |
| The registered office of IR-Med, Inc. and the corporate headquarters and research facility of IR. Med, Ltd. are located in Rosh Pina, Israel. The Parent Company and the Subsidiary are at times collectively referred to as the “Company”. | |
| The Company is a development stage medical device company developing its technology through its Subsidiary and is utilizing Infra-Red-light spectroscopy (IR) combined with Artificial Intelligence (AI) technology platform to develop non-invasive devices for various medical indications, by detecting and measuring various biomarkers and molecules in the blood and in human tissue in real-time. The initial product candidates which are currently in various stages of development are non-invasive, user friendly and designed to address the medical needs of large and growing target patient groups by offering earlier and more accurate information for detection, which is expected to reduce healthcare expenses and reducing the widespread reliance on antibiotics administration, and other interventional options optimizing the delivery of the targeted medical services and, as a result, improving the efficacy and safety of administered treatments. |
| B. | Going Concern |
| The
Company is in its development stage and does not expect to generate significant revenue until such time as the Company shall have
completed the design and development of its initial product candidate and obtained the requisite approvals to market the product.
During the year ended December 31, 2023, the Company has incurred losses of US$ | |
| Management’s plans regarding these matters include continued development and marketing of the Company’s products, as well as seeking additional financing arrangements. Although management continues to pursue these plans, there is no assurance that the Company will be successful in raising the needed capital from revenues or financing on commercially acceptable terms. The Company will require additional liquidity to continue its operations over the next 12 months. | |
| Following the brutal attacks on Israel, the mobilization of army reserves and the Israeli Government declaring a state of war (the “Iron Swords War”) in October 2023, there has been a decrease in Israel’s economic and business activity. The security situation has led, inter alia, to a disruption in the chain of supply and production, a decrease in the volume of national transportation, a shortage in manpower as well as a decrease in the value of financial assets and a rise in the exchange rate of foreign currencies in relation to the shekel. |
| F-9 |
IR-Med, Inc.
Notes to the Consolidated Financial Statements
Note 1 - General (Cont’d)
| B. | Going Concern (cont’d) |
| At this time, the Company has assessed, on the basis of the information it has as of the date of the approval of these financial statements, that the current events and the escalation in security in Israel, may have a material effect on the business plans of the Company in the short term. As a result of the movement and work restrictions in Israel, the Company has begun operating on a limited scale. These restrictions and the shortage in manpower may cause delays in the Company’s research and development activities and in its marketing efforts. In addition, the situation has brought further difficulties in management’s efforts to seek additional financing arrangements. Since this is an event that is not under the control of the Company, and matters such as the fighting continuing or stopping may affect the Company’s assessments, as at the reporting date the Company is unable to assess the extent of the effect of the Iron Swords War on its business | |
| On
June 12, 2023, the Company entered into a subscription agreement with one investor pursuant to which the Company issued
shares of its common stock at a per share price of $ and warrants to purchase up to an additional | |
| As a result of the Company’s financial condition substantial doubt exists that the Company will be able to continue as a going concern for one year from the issuance date of this 2023 Annual Report. The consolidated financial statements do not include any adjustments to the carrying amounts and classification of assets, liabilities, and reported expenses that may be necessary if the Company is unable to continue as a going concern. |
Note 2 - Summary of Significant Accounting Policies
A. Basis of Presentation
| The financial statements have been prepared in accordance with U.S. generally accepted accounting principles (“U.S. GAAP”). These consolidated financial statements have been prepared in accordance with U.S. generally accepted accounting principles (GAAP) assuming the Company will continue as a going concern. The going concern assumption contemplates the realization of assets and satisfaction of liabilities in the normal course of business. However, substantial doubt about the Company’s ability to continue as a going concern exists. |
| F-10 |
IR-Med, Inc.
Notes to the Consolidated Financial Statements
Note 2 - Summary of Significant Accounting Policies (Cont’d)
B. Functional Currency
| The Company finances its operations in U.S. dollars. While the majority of the Group’s operations are currently conducted in Israel, a significant part of the Company’s expenses is denominated and determined in U.S. dollars Future revenues are expected to be earned in US dollars. The Company’s management believes that the U.S. Dollar is the currency of the primary economic environment in which the Company operates and expects to continue to operate in the foreseeable future. Thus, the functional and reporting currency of the Company is the U.S. Dollar. | |
| The Company’s transactions and balances denominated in U.S. dollars are presented at their original amounts. Non-Dollar transactions and balances have been re-measured to U.S. Dollars in accordance with Accounting Standards Codification (ASC) 830, “Foreign Currency Matters”, of the Financial Accounting Standards Board (“FASB”). All transaction gains and losses from re-measurement of monetary balance sheet items denominated in non-dollar currencies are reflected in the statements of operations as financial income or expenses, as appropriate. |
C. Principles of Consolidation
| The consolidated financial statements include the accounts of the Parent Company and its wholly owned Subsidiary, IR. Med, Ltd. Intercompany transactions and balances have been eliminated in consolidation. |
D. Use of Estimates
| The preparation of financial statements in conformity with U.S. GAAP requires management to make judgments, estimates and assumptions that affect the reported amounts of assets and liabilities and disclosures of contingent assets and liabilities at the date of the consolidated financial statements, and the reported amounts of expenses during the reporting period. Significant items subject to such estimates and assumptions including fair value of share-based compensation and legal claims. Actual results could differ from those estimates. |
E. Cash and Cash Equivalents
| The Group considers all highly liquid investments with a maturity of three months or less at the date of purchase to be cash equivalents. Cash equivalents are stated at their carrying values, which approximates their fair values. |
F. Property and Equipment
| Property and equipment are stated at cost less accumulated depreciation and accumulated impairment losses, if any. Maintenance and repair expenses are charged to operation as incurred. Depreciation is calculated on the straight-line method based on the estimated useful lives of the assets and commences once the assets are ready for their intended use. The cost of property and equipment include expenditure that is attributable to the acquisition of the assets. |
| F-11 |
IR-Med, Inc.
Notes to the Consolidated Financial Statements
Note 2 - Summary of Significant Accounting Policies (Cont’d)
Annual rates at depreciation are as follows:
| % | ||||
| Computers and electronics equipment | ||||
| Laboratory equipment | ||||
| Furniture and equipment | ||||
| Leasehold improvements | ||||
G. Research and Development Expenses
| Research and development expenses are expensed as incurred. Those expenses include payments to third party consultants, expenses related to conducting clinical and pre-clinical trials, salaries and related personnel expenses. |
H. Fair Value of Financial Instruments
| The carrying amounts of the Company’s financial instruments, including cash and cash equivalents, accounts receivables, trade and other accounts payable and stockholders’ loans do not significantly vary from their fair values. Amounts from related parties’ approximate fair value because of their short-term nature. | |
| Fair value for the measurement of financial assets and liabilities is defined as the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. As such, fair value is a market-based measurement that should be determined based on assumptions that market participants would use in pricing an asset or liability. The Group utilizes a valuation hierarchy for disclosure of the inputs for fair value measurement. This hierarchy prioritizes the inputs into three broad levels as follows: |
| ● | Level 1 inputs are unadjusted quoted prices in active markets for identical assets or liabilities. |
| F-12 |
IR-Med, Inc.
Notes to the Consolidated Financial Statements
Note 2 - Summary of Significant Accounting Policies (Cont’d)
| H. | Fair Value of Financial Instruments (cont’d) |
| ● | Level 2 inputs are quoted prices for identical or similar assets or liabilities in less active markets or model derived valuations in which significant inputs are observable for the asset or liability, either directly or indirectly through market corroboration. | |
| ● | Level 3 inputs are unobservable inputs based on the Company’s assumptions used to measure assets and liabilities at fair value. |
| By distinguishing between inputs that are observable in the marketplace, and therefore more objective, and those that are unobservable and therefore more subjective, the hierarchy is designed to indicate the relative reliability of the fair value measurements. A financial asset or liability’s classification within the hierarchy is determined based on the lowest level input that is significant to the fair value measurement. |
I. Warrants
| The Company assesses whether warrants issued require accounting as derivatives. The Company determined that the warrants were (1) indexed to the Company’s own stock and (2) classified in stockholders’ equity in accordance with FASB ASC Topic 815, Derivatives and Hedging. As such the Company has concluded the warrants meet the scope exception for determining whether the instruments require accounting as derivatives and should be classified in stockholders’ equity |
J. Commitments and Contingencies
| Liabilities for loss contingencies arising from claims, assessments, litigations, fines and penalties and other sources are recognized when it is probable that a liability has been incurred and the amount of the assessment can be reasonably estimated. |
| Stock Option Plan | |
| The Group recognizes all employee and nonemployee stock-based compensation as a cost in the consolidated financial statements. For awards with a graded vesting schedule, the Company uses the graded vesting attribution approach to recognize compensation cost over the vesting period. | |
| The Group estimates grant date fair value using the Black-Scholes-Merton option-pricing model and estimates the number of forfeitures expected to occur. |
L. Income taxes
| Income taxes are accounted for under the asset and liability method. Deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statements carrying amounts of existing assets and liabilities and their respective tax bases and operating loss and tax credit carryforwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period that includes the enactment date. The Group records valuation allowances to reduce deferred income tax assets to the amount that is more likely than not to be realized. |
| F-13 |
IR-Med, Inc.
Notes to the Consolidated Financial Statements
Note 2 - Summary of Significant Accounting Policies (Cont’d)
L. Income taxes(cont’d)
| The Group recognizes the effect of income tax positions only if those positions are more likely than not of being sustained. Recognized income tax positions are measured at the largest amount that is greater than 50% likely of being realized. Changes in recognition or measurement are reflected in the period in which the change in judgment occurs. The Company accounts for interest and penalties related to an underpayment of income taxes as a component of income tax expense. |
M. Concentrations of credit risks
| Financial instruments that potentially subject the Group to concentrations of credit risk consist principally of cash and cash equivalents. Cash and cash equivalents are held in commercial banks in the U.S. and in Israel. Management believes that the financial institution that holds the Group investments have high credit ratings. The Group has no off-balance-sheet concentration of credit risk such as foreign exchange contracts, option contracts or other foreign hedging arrangements. |
N. Employee benefits
| Pension | |
| The Company’s liability for severance pay for its Israeli employees is calculated pursuant to Israeli severance pay law based on the most recent salary of the employee multiplied by the number of years of employment, as of the balance sheet date. Those employees are entitled to one month’s salary for each year of employment or a portion thereof. | |
| The Group has a defined deposit plan in respect of the Company’s obligation to pay the benefit component of provident funds as well as in respect of its employees to whom section 14 of the Dismissal Compensation Law applies. pursuant to the terms of Section 14 of the Israeli Severance Pay Law, 1963, according to which the current deposits in the pension fund and/or with the insurance company exempt the Company from any additional obligation to these employees for whom the said depository payments are made. During the years ended December 31, 2023, and 2022, the Company paid for severance pay for its Israeli employees amounts of NIS 415 thousand (approximately US$113 thousand) and NIS 461 thousand (approximately US$144 thousand), respectively. |
| Short-term benefits | |
| Short-term employee benefit obligations are measured on an undiscounted basis and are expensed as the related service is provided or upon the actual absence of the employee when the benefit is not accumulated (such as maternity leave). | |
| The employee benefits are classified, for measurement purposes, as short-term benefits or as other long-term benefits depending on when the Group expects the benefits to be wholly settled. |
O. Governments grants
| The Company records grants received from the Israel Innovation Authority (the “IIA”, formerly known as the Office of the Chief Scientist of the Israeli Ministry of Industry and Trade) as a liability, if it is probable that the Company will have to repay the grants received. If it is not probable that the grants will be repaid, the Company records the grants as a reduction to research and development expenses. Royalties paid to the IIA are recognized as a reduction of the above-mentioned liability. |
| F-14 |
IR-Med, Inc.
Notes to the Consolidated Financial Statements
Note 2 - Summary of Significant Accounting Policies (Cont’d)
P. Leases
| The Subsidiary is a lessee in several noncancellable operating leases, primarily for transportation. The Company accounts for leases in accordance with Topic 842, Leases. The Company determines if an arrangement is or contains a lease at contract inception. The Company recognizes a right-of-use (ROU) asset and a lease liability at the lease commencement date. For operating leases, the lease liability is initially and subsequently measured at the present value of the unpaid lease payments at the lease commencement date. | |
| Key estimates and judgments include how the Company determines (1) the discount rate it uses to discount the unpaid lease payments to present value, (2) lease term and (3) lease payments. | |
| Operating leases are included in ROU assets and trade and other payables in the consolidated balance sheet. | |
| Topic 842 requires a lessee to discount its unpaid lease payments using the interest rate implicit in the lease or, if that rate cannot be readily determined, its incremental borrowing rate. Generally, the Company cannot determine the interest rate implicit in the lease because it does not have access to the lessor’s estimated residual value or the amount of the lessor’s deferred initial direct costs. Therefore, the Company generally uses its incremental borrowing rate as the discount rate for the lease. The Company’s incremental borrowing rate for a lease is the rate of interest it would have to pay on a collateralized basis to borrow an amount equal to the lease payments under similar terms. Because the Company does not generally borrow on a collateralized basis, it uses the interest rate it pays on its noncollateralized borrowings as an input to deriving an appropriate incremental borrowing rate, adjusted for the amount of the lease payments, the lease term, and the effect on that rate of designating specific collateral with a value equal to the unpaid lease payments for that lease. | |
| Our subsidiary occupies approximately |
| Lease payments included in the measurement of the lease liability comprise of the following: |
| — | Fixed payments, including in-substance fixed payments, owed over the lease term (which includes termination penalties the Company would owe if the lease term assumed the Company’s exercise of a termination option); | |
| — | Variable lease payments that depend on an index or rate, initially measured using the index or rate at the lease commencement date. |
| The Right of Use (ROU) asset is initially measured at cost, which comprises the initial amount of the lease liability adjusted for lease payments made at or before the lease commencement date, plus any initial direct costs incurred less any lease incentives received. |
| F-15 |
IR-Med, Inc.
Notes to the Consolidated Financial Statements
Note 2 - Summary of Significant Accounting Policies (Cont’d)
P. Leases (Cont’d)
| For operating leases, the ROU asset is subsequently measured throughout the lease term at the carrying amount of the lease liability, plus initial direct costs, plus (minus) any prepaid (accrued) lease payments, less the unamortized balance of lease incentives received. Lease expense for lease payments is recognized on a straight-line basis over the lease term. | |
| Variable lease payments associated with the Company’s leases are recognized when the event, activity, or circumstance in the lease agreement on which those payments are assessed occurs. Variable lease payments are presented as operating expense in the Company’s consolidated statements of income in the same line item as expense arising from fixed lease payments (operating leases) or amortization of the ROU asset (finance leases). |
The Company uses the long-lived assets impairment guidance in Accounting Standards Codification (“ASC”) Subtopic 360-10, Property, Plant, and Equipment – Overall, to determine whether an ROU asset is impaired, and if so, the amount of the impairment loss to recognize. The Company monitors for events or changes in circumstances that require a reassessment of one of its leases. When a reassessment results in the remeasurement of a lease liability, a corresponding adjustment is made to the carrying amount of the corresponding ROU asset unless doing so would reduce the carrying amount of the ROU asset to an amount less than zero. In that case, the amount of the adjustment that would result in a negative ROU asset balance is recorded in profit or loss. Operating lease ROU assets are presented as operating lease right of use assets on the consolidated balance sheet. The current portion of operating lease liabilities is included in trade and other payables and the long-term portion is presented separately as operating lease liabilities on the consolidated balance sheet.
| The Company recognizes the lease payments associated with its leases of motor vehicles as an expense on a straight-line basis over the lease term. Variable lease payments associated with these leases are recognized and presented in the same manner as for all other Company leases. |
Q. Stockholders’ loans
| Shareholders’ loans with terms that were amended after the reporting date are considered in determining the classification of debt at the reporting date. Due to agreements reached in 2024 between the shareholders and the Company regarding the repayment date of the loan (see note 8), the shareholders’ loans at December 31, 2023 are classified as non-current liabilities. |
| F-16 |
IR-Med, Inc.
Notes to the Consolidated Financial Statements
R. New Accounting Pronouncements
In December 2023, the FASB issued Accounting Standards Update No. 2023-09, Income Taxes (Topic 740) Improvements to Income Tax Disclosure. The standard requires to disclose additional information in tax rate reconciliation table about federal, state and foreign income taxes and to provide more details about the reconciling items in some categories. The standard will become effective for fiscal years beginning after December 15, 2024. The Company is currently assessing the impact of the adoption of this standard on its consolidated financial statements.
Note 3 - Cash and Cash Equivalents
| December 31 | December 31 | |||||||
| 2023 | 2022 | |||||||
| US Dollars (In thousands) | ||||||||
| Cash - NIS | ||||||||
| Cash - US dollars | ||||||||
Note 4 - Accounts Receivable
| December 31 | December 31 | |||||||
| 2023 | 2022 | |||||||
| US Dollars (In thousands) | ||||||||
| Prepaid expenses | ||||||||
| Government institutions | ||||||||
| Related parties | ||||||||
Note 5 – Property and Equipment, net
| December 31 | December 31 | |||||||
| 2023 | 2022 | |||||||
| US Dollars (In thousands) | ||||||||
| Computers and electronics’ equipment | ||||||||
| Laboratory equipment | ||||||||
| Furniture and equipment | ||||||||
| Leasehold improvement | ||||||||
| Less – accumulated depreciation | ( | ) | ( | ) | ||||
| F-17 |
IR-Med, Inc.
Notes to the Consolidated Financial Statements
Note 6 - Leases
The
Company has six lease contracts for motor vehicles used in its operation. Leases of motor vehicles generally have lease terms of
The
above operating leases are included in “Right-of-use assets” on the Company’s Consolidated Balance Sheets as of December
31, 2023, and represent the Company’s right to use the underlying asset for the lease term. The Company’s obligations to
pay lease payments are included as “Lease liability” on the Company’s Consolidated Balance
Sheets as of December 31, 2023. Based on the present value of the lease payments for the remaining lease term of the Company’s
existing lease agreements, the Company recognized operating right-of-use assets and operating lease liabilities of approximately US$
During
the years ended December 31, 2023, and 2022, the Company recognized expenses of US$
During
the years ended December 31, 2023, and 2022, the Company recognized a decrease in right-of-use assets of US$
Maturities of lease liabilities under noncancellable leases as of December 31, 2023, are as follows: (US dollars in thousands):
| 2024 | ||||
| 2025 | ||||
| Total future minimum lease payments | ||||
| Less imputed interest: | ( | ) | ||
| Present value of operating lease liabilities |
| F-18 |
IR-Med, Inc.
Notes to the Consolidated Financial Statements
Note 7 - Trade and Other Payables
| December 31 | December 31 | |||||||
| 2023 | 2022 | |||||||
| US Dollars (In thousands) | ||||||||
| Trade payables | ||||||||
| Accrued expenses | ||||||||
| Payroll and related accruals | ||||||||
| Advance from customer | ||||||||
| Lease liability | ||||||||
| Related Parties | ||||||||
| Other | ||||||||
Note 8 - Stockholders’ Loans
| A. | In
2015, certain of the Company’s stockholders granted loans to the Company to finance its ongoing operation (hereinafter: the “2015
Loans”). These loans bear interest at an annual rate ranging in 2023 and 2022 from |
| In March 2020, the Company and the lender agreed to amend the terms of the 2015 Loan and the repayment date was extended to December 31, 2023. On March 1, 2024, the Company and the lender agreed to extend the repayment date to December 31, 2025. | |
| As
of December 31, 2023, and 2022, the carrying amounts of the 2015 Loans were US$ | |
| In
2017, one of the Company’s shareholders provided the Company with a loan to finance its ongoing operation (hereinafter: the
“2017 Loan”). This loan bears interest at annual rate ranging in 2023 and 2022 from | |
| In
March 2020, the Company and the lender agreed to amend the terms of the 2017 Loan and the repayment date was extended to December
31, 2023. On March 1, 2024, the Company and the lender agreed to extend the repayment date to | |
| As
of December 31, 2023, and 2022, the carrying amounts of the 2017 Loan were US$ |
| F-19 |
IR-Med, Inc.
Notes to the Consolidated Financial Statements
Note 8 - Stockholders’ Loans (cont’d)
| B. | Convertible Loan |
| On March 6, 2018, certain of the Company’s shareholders entered with the Company into a convertible bridge loan agreement (the “2018 CLA”). | |
| The CLA included certain scenarios in which the loan may be converted (“Optional conversion”), and certain scenarios in which the loan is automatically converted (“Mandatory conversion”). | |
| The Company recorded the loan amount as a liability, applying the accounting guidance in ASC 835-30. The embedded derivatives identified by the Company relating to the Exit event and Optional conversion were estimated by the Company as immaterial amounts. | |
| In late 2018, the Majority Lenders agreed to defer the repayment date of the loan to a later date, after December 31, 2019. During 2018 and 2019 the convertible loan was not converted into shares. | |
| In
March 2020, the Company and the lenders agreed to amend and restate the 2018 CLA (“the Amended CLA”) pursuant to which
the lenders waived any and all rights to convert their respective outstanding loan amounts, and the repayment date was set to | |
| On March 1, 2024, the Company and the lenders agreed to extend the repayment date to December 31, 2025. | |
| Financing
expenses recorded in respect of the loan during 2023 and 2022 were US$ | |
| As
of December 31, 2023, and 2022, the carrying amounts of the loans were US$ |
| F-20 |
IR-Med, Inc.
Notes to the Consolidated Financial Statements
Note 9 - Stockholders’ Equity
| A. | Common Stock |
| As of December 31, 2023, the Company has shares of Common Stock issued and outstanding. | |
| Each share of IR-Med, Inc.´s common stock entitles its holder to one vote, and all shares rank equally as to voting and other matters. | |
| Dividends may be declared and paid on the common stock from funds legally available therefor, if, as and when determined by the Board of Directors. |
| B. | Financing round |
| (I) | During
the first four months of 2021, the Company raised in the aggregate an additional $ | |
| (II) | Between
April 2022 through July 2022, the Company entered into a securities purchase agreement with six accredited investors providing for
the issuance and sale to such investors of an aggregate of shares of the Company common stock and warrants for an additional
shares of the Company common stock, exercisable through 2024, at a per share exercise price of $ | |
| (III) | On
June 12, 2023, the Company entered into a subscription agreement with one investor pursuant to which the Company issued
shares of its common stock at a per share price of $ and warrants to purchase up to an additional |
| F-21 |
IR-Med, Inc.
Notes to the Consolidated Financial Statements
Note 9 - Stockholders’ Equity (cont’d)
| C. | Share-based compensation |
| On December 23, 2020, the Group’s board of directors approved, and the shareholders adopted a share-based compensation plan (“2020 Incentive Stock Plan”) for future grants by the Parent Company. On September 27, 2023, our Board approved a further amendment to the 2020 Incentive Stock Plan to increase the number of shares authorized for issuance of awards under the 2020 Incentive Stock Plan from shares to an aggregate of shares of common stock. The holders of a majority of our voting stock approved such an increase. | |
| As of December 31, 2023,
the Parent Company awarded to its employees and service providers options to purchase up to
shares of common stock, of which options for
shares were at an exercise price of US$
per share, options for
shares were at an exercise price of US$
per share, options for
shares were at an exercise price of US$
per share and options for
shares were are an exercise price of US$
per share. As of December 31, 2023, options for
shares were vested with weighted average of exercise price of US$ , and the remaining balance has a vesting period ranging
between one to five years. The options are exercisable for periods ranging between
to | |
| Options awarded: |
| 2023 | ||||||||
| Weighted average of exercise price | Number of options | |||||||
| Outstanding as of beginning of year | $ | |||||||
| Granted | $ | |||||||
| Cancelled | $ | ( | ) | |||||
| Outstanding as of end of year | $ | |||||||
On June 12, 2022, the Parent Company entered into a consulting agreement for professional services. As part of this agreement, the parent company granted the consultant shares at par value per month. As of December 31, 2023, the parent company granted the consultant shares at par value.
On August 15, 2022, the Parent Company entered into a consulting agreement for professional services. As part of this agreement, the parent company granted the consultant shares at par value.
| F-22 |
IR-Med, Inc.
Notes to the Consolidated Financial Statements
Note 9 - Stockholders’ Equity (cont’d)
| C. | Share-based compensation (cont’d) |
| On June 1, 2023, the Parent Company entered into a consulting agreement for professional services. As part of this agreement, the Parent Company will grant the consultant shares at par value in four tranches. As of December 31, 2023, the Parent Company granted the consultant shares at par value. | |
| The
grant was approved following the adoption of the 2020 Incentive Stock Plan by the Parent Company on December 23, 2020, and the
adoption of the sub plan (the “Israeli appendix”) on April 29, 2021. The Group recorded in the statement of operations
a non-cash expense of US$ | |
| The stock-based compensation expenses for the years ended December 31, 2023, and 2022 were recognized in the statements of operations as follows; |
| Year ended December 31 | ||||||||
| 2023 | 2022 | |||||||
| US Dollars (In thousands) | ||||||||
| Research and development expenses | ||||||||
| Marketing expenses | ||||||||
| General and administrative expenses | ||||||||
The Company will not be allowed to claim as an expense for tax purposes the amounts charged as stock-based compensation expenses.
The following table sets forth information about the weighted-average fair value of options granted to employees and service providers during the year period ended December 31, 2023, using the Black-Scholes-Merton option-pricing model and the weighted-average assumptions used for such grants:
| F-23 |
IR-Med, Inc.
Notes to the Consolidated Financial Statements
Note 9 - Stockholders’ Equity (cont’d)
| C. | Share-based compensation (cont’d) |
| For the years ended | ||||||||
| December 31 | December 31 | |||||||
| 2023 | 2022 | |||||||
| Dividend yields (see (I) below) | % | % | ||||||
| Share price (in U.S. dollar) (see (II) below) | , | , | ||||||
| Expected volatility (see (III) below) | %- | % | %- | % | ||||
| Risk-free interest rates (see (IV) below) | %- | % | %- | % | ||||
| Expected life (in years) (see (V) below) | - | - | ||||||
| I. | ||
| II. | ||
| III. | ||
| IV. | ||
| V. |
| F-24 |
IR-Med, Inc.
Notes to the Consolidated Financial Statements
Note 10 - Contingent Liabilities and Commitments
| A. | Israel Innovation Authority |
| The
Company operates within the framework of the Incubators Program (Directive No. 8.3 of the Ministry of Economy) (“The program”).
As part of this plan, | |
| As
of December 31, 2023, the Company’s maximum possible future royalties commitment, subject to future sales of such products
and based on grants received from the IIA and not yet repaid, is approximately $ | |
| For the years ending December 31, 2023, and 2022, no additional IIA grants were obtained. |
| B. | Vehicle Leases |
| The
Company has lease contracts for six motor vehicles used in its operations in Israel. Motor vehicle leases generally have lease terms
of three years and require a deposit amount of three-monthly lease payment. As of December 31, 2023, the Company deposited an
aggregate of NIS |
| C. | Long term deposits |
| During
2021 the Company received a bank credit line in the amount of NIS | |
| (Approximately
US$ |
| D. | Legal claims |
| On
May 29, 2023, a lawsuit was filed against the Company, the Subsidiary and Mr. Aharon Klein (the “Plaintiff”), a Company Director and the
Company’s Chief Technology Officer in the Tel Aviv District Court of Israel, by an individual who provided, on part time
basis, certain consulting services to the Subsidiary between October 2015 and October 2016, before the acquisition of the
Subsidiary by the Company. The suit alleges breach of contract by the defendants based on non-payment of amounts purportedly owed to
the Plaintiff in respect of the services rendered, including the market value of the Company’s common stock that the Plaintiff
alleges should have been issued to him in respect of services. The suit seeks declaratory judgment that the defendants breached
certain agreements with the Plaintiff and claimed damages in the aggregate amount of approximately $ | |
| The Company records a provision in its financial statements to the extent that it concludes that a contingent liability is probable, and the amount thereof is reasonably estimable. Based upon the status of the case described above, management’s assessments of the likelihood of damages, and the advice of counsel, no provisions have been made regarding the matter disclosed in this note. Litigation outcomes and contingencies are unpredictable, and excessive verdicts can occur. | |
| Accordingly, management’s assessments involve complex judgments about future events and often rely heavily on estimates and assumptions. |
| F-25 |
IR-Med, Inc.
Notes to the Consolidated Financial Statements
Note 11 - Research and Development Expenses
| For the years ended | ||||||||
| December 31 | December 31 | |||||||
| 2023 | 2022 | |||||||
| US Dollars (In thousands) | ||||||||
| Salaries and related expenses | ||||||||
| Subcontractors | ||||||||
| Materials | ||||||||
| Stock based compensation expenses | ||||||||
| Other expenses | ||||||||
| Total research and development expenses | ||||||||
Note 12 - Marketing Expenses
| For the years ended | ||||||||
| December 31 | December 31 | |||||||
| 2023 | 2022 | |||||||
| US Dollars (In thousands) | ||||||||
| Salaries and related expenses | ||||||||
| Professional expenses | ||||||||
| Other expenses | ||||||||
| Stock based compensation expenses | ||||||||
| Total marketing expenses | ||||||||
| F-26 |
IR-Med, Inc.
Notes to the Consolidated Financial Statements
Note 13 - General and Administrative Expenses
| For the years ended | ||||||||
| December 31 | December 31 | |||||||
| 2023 | 2022 | |||||||
| US Dollars (In thousands) | ||||||||
| Salaries and related expenses | ||||||||
| Professional expenses | ||||||||
| Rent and Maintenance | ||||||||
| Depreciation | ||||||||
| Other expenses | ||||||||
| Stock based compensation expenses | ||||||||
| Total general and administrative expenses | ||||||||
Note 14 - Financial Expenses
| For the years ended | ||||||||
| December 31 | December 31 | |||||||
| 2023 | 2022 | |||||||
| US Dollars (In thousands) | ||||||||
| Interest (income) expenses, net | ( | ) | ||||||
| Exchange rate loss (gain) | ( | ) | ||||||
| Other | ||||||||
| Total financial income, net | ( | ) | ( | ) | ||||
| F-27 |
IR-Med, Inc.
Notes to the Consolidated Financial Statements
| For the years ended | ||||||||
| December 31 2023 | December 31 2022 | |||||||
| US Dollars (In thousands) | ||||||||
| Loss attributable to shareholders ($ in thousands) | ( | ) | ( | ) | ||||
| Weighted average number of ordinary shares: | ||||||||
| Balance at beginning of year | ||||||||
| Effect of shares issued during the year | ||||||||
| Weighted-average shares – basic and dilutive as at end of year | ||||||||
| Basic and dilutive loss per share ($) | ) | ) | ||||||
As of December 31, 2023, and 2022, options and warrants which were granted by the Group’s board of directors were not included in the loss per share computation because all such securities have an anti-dilutive effect for the year. Such securities totaled to and , respectively.
Note 16 – Income Taxes
| A. | Corporate income tax rate |
| a) | The
income tax rates relevant to the Parent company in Nevada for the years 2023 and 2022 was | |
| The
tax rates relevant to the Subsidiary for the years 2023 and 2022 was | ||
| Current taxes for the reported periods are calculated according to the enacted tax rates presented above. | ||
| b) | Tax benefits under the Israeli Law for the Encouragement of Capital Investments, 1959 (the “Investments Law”). | |
| The Israeli Investments Law applies to Preferred Income derived or accrued in 2011 and thereafter by a Preferred Company. |
| F-28 |
IR-Med, Inc.
Notes to the Consolidated Financial Statements
Note 16 – Income Taxes (Cont’d)
The
law provides a uniform and reduced income tax rate for all the Subsidiary’s income entitled to the benefits (“Preferred Income”).
Starting from tax year 2017, the tax rate on Preferred Income for a company operating in the same area as the Subsidiary is
| B. | Deferred tax assets |
The following is a summary of the significant components of deferred tax assets:
| December 31 | December 31 | |||||||
| 2023 | 2022 | |||||||
| US Dollars (In thousands) | ||||||||
| Operating loss carryforwards | ||||||||
| Research and development costs capitalized for tax purposes | ||||||||
| Payroll and related payables | ||||||||
| Lease liability | ||||||||
| Total deferred tax assets | ||||||||
| Right of use asset- deferred tax liability | ( | ) | ( | ) | ||||
| Total deferred tax assets, net | ||||||||
| Valuation allowance for deferred tax assets | ( | ) | ( | ) | ||||
| Deferred tax assets, net of valuation allowance | ||||||||
As
of December 31, 2023, and 2022, the Company has provided full valuation allowance of US$
| C. | Accounting for uncertainty in income taxes |
| For the years ended December 31, 2023, 2022, the Company did not have any significant unrecognized tax benefits. In addition, the Company does not expect that the amount of unrecognized tax benefits will change significantly within the next 12 months. | |
| The
Company accounts for interest and penalties related to an underpayment of income taxes as a component of income tax expense. For the
years ended December 31, 2023 and 2022, |
| D. | Operating losses carry forwards and valuation allowance |
| As
of December 31, 2023, and 2022, the Company had operating loss carryforwards in the amount of US$ |
| F-29 |
IR-Med, Inc.
Notes to the Consolidated Financial Statements
Note 16 – Income Taxes (Cont’d)
| E. | Composition of loss from continuing operations before income taxes: |
| December 31 | December 31 | |||||||
| 2023 | 2022 | |||||||
| US Dollars (In thousands) | ||||||||
| Loss from continuing operations before income taxes: | ||||||||
| US | ||||||||
| Israel | ||||||||
| F. | Income tax assessment |
| As of December 31, 2023, the Subsidiary has tax assessments that are considered as final in Israel due to lapse of statute of limitation period, through tax year 2018. The Parent Company has not been assessed for income tax purposes in the U.S. since its inception and is open to examination by the IRS from the tax year 2019. |
| G. | Reconciliation of the statutory income tax expense (benefit) to actual income tax expense |
| For the years ended | ||||||||
| December 31 | December 31 | |||||||
| 2023 | 2022 | |||||||
| US Dollars (In thousands) | ||||||||
| Loss before income taxes as reported in the statements of operations | ( | ) | ( | ) | ||||
| Statutory tax rate | % | % | ||||||
| Theoretical income tax benefit on the above amount at the US federal statutory income tax rate | ( | ) | ( | ) | ||||
| F-30 |
IR-Med, Inc.
Notes to the Consolidated Financial Statements
Note 16 – Income Taxes (Cont’d)
| G. | Reconciliation of the statutory income tax expense (benefit) to actual income tax expense (cont’d) |
| Additional tax (tax savings) in respect of: | ||||||||
| Nondeductible expenses | ||||||||
| Differences in tax rates between statutory tax and income tax of the Subsidiary* | ( | ) | ( | ) | ||||
| Change in valuation allowance | ||||||||
| Effect of currency exchange differences | ||||||||
| Other | ||||||||
| Actual taxes on income |
| (*) |
| H. | Roll forward of valuation allowance |
| US Dollars (In thousands) | ||||
| Balance on January 1, 2022 | ||||
| Change in valuation allowance – income tax expenses | ||||
| Balance on December 31, 2022 | ||||
| Change in valuation allowances - Income tax expense | ||||
| Balance on December 31, 2023 | ||||
| F-31 |
IR-Med, Inc.
Notes to the Consolidated Financial Statements
Note 17 - Related Parties Balances and Transactions
The Group’s related parties are seven directors, four officers, one shareholder and two entities controlled by three of the Company’s shareholders.
| A. | Balances with related parties |
| December 31 | December 31 | |||||||
| 2023 | 2022 | |||||||
| US Dollars (In thousands) | ||||||||
| Assets | ||||||||
| Other receivables | ||||||||
| Liabilities | ||||||||
| Payables | ||||||||
| Accrued expenses | ||||||||
| Payroll and related | ||||||||
| Stockholders’ loans | ||||||||
| B. | Transactions with related parties |
| For the years ended | ||||||||
| December 31 | December 31 | |||||||
| 2023 | 2022 | |||||||
| US Dollars (In thousands) | ||||||||
| Subcontractors and professional expenses (1) | ||||||||
| Salaries and related expenses (2) | ||||||||
| Stock based compensation (3) | ||||||||
| Rent and Maintenance (4) | ||||||||
| Interest expenses, see note 8 | ||||||||
| (1) | ||
| For the years ended December 31, 2023, and 2022, the Company paid to one shareholder of the Parent Company and his relative an aggregate consideration of |
| F-32 |
IR-Med, Inc.
Notes to the Consolidated Financial Statements
Note 17 - Related Parties Balances and Transactions (Cont’d)
| B. | Transactions with related parties (cont’d) |
| US$ | |
| For
the years ended December 31, 2023, and 2022, the Company paid to the Parent Company non-employee directors an aggregate consideration
of US$ |
| (2) | ||
| For
the year ended December 31, 2022, the Company paid to one director of the Parent Company and four of its officers, salary and related
expenses totaled to US$ | ||
| (3) | ||
| The Parent Company granted during the year 2022 to its directors, officers and a shareholder options to purchase shares of Common Stock (See also note 9-C). | ||
| (4) |
Note 18 – Subsequent Events
During
February 2024, as a result of financial difficulties, the Company notified 10 of its
On
January 25, 2024, the Israel Innovation Authority (the “IIA”) approved a program to develop a device for the early detection
of diabetic foot ulcers among diabetic patients, with a project budget of NIS
| F-33 |
IR-MED, INC.
Form 10-Q
March 31, 2024
| F-34 |
IR-Med
Inc.
Interim Unaudited Condensed Consolidated Balance Sheets
| March 31 2024 | December 31 2023 | |||||||
| USD thousands | USD thousands | |||||||
| Assets | ||||||||
| Current assets | ||||||||
| Cash and cash equivalents | ||||||||
| Accounts receivable | ||||||||
| Total current assets | ||||||||
| Non- current assets | ||||||||
| Long term restricted deposit | ||||||||
| Right of use assets | ||||||||
| Property and equipment, net | ||||||||
| Total non-current assets | ||||||||
| Total assets | ||||||||
| Liabilities and stockholders’ equity (deficiency) | ||||||||
| Current liabilities | ||||||||
| Trade and other payables | ||||||||
| Non-current liabilities | ||||||||
| Stockholders’ loans | ||||||||
| Total liabilities | ||||||||
| Stockholders’ equity (deficiency) | ||||||||
| Common Stock, par value $ per share, , shares authorized. As of March 31, 2024, and December 31, 2023, shares were issued. | ||||||||
| Additional paid-in capital | ||||||||
| Accumulated deficit | ( | ) | ( | ) | ||||
| Total Stockholders’ equity (deficiency) | ( | ) | ||||||
| Total liabilities and stockholders’ equity (deficiency) | ||||||||
The accompanying notes are an integral part of these interim unaudited condensed consolidated financial statements.
| F-35 |
IR-Med Inc.
Interim Unaudited Condensed Consolidated Statements of Operations
For the three-months period ended March 31 | ||||||||
| 2024 | 2023 | |||||||
| U.S dollars (in thousands) | ||||||||
| Research and development expenses: | ||||||||
| Expenses incurred | ||||||||
| Less- government participation | ( | ) | ||||||
| Research and development expenses, net | ||||||||
| Marketing expenses | ||||||||
| General and administrative expenses | ||||||||
| Total operating loss | ||||||||
| Financial income, net | ( | ) | ( | ) | ||||
| Loss for the period | ||||||||
| Basic and dilutive loss per common stock (in dollars) | ) | ) | ||||||
Weighted-average shares in the loss per share computation for the three months ended March 31, 2024 and, 2023 were and respectively.
The accompanying notes are an integral part of these interim unaudited condensed consolidated financial statements.
| F-36 |
IR-Med
Inc.
Interim Unaudited Condensed Consolidated Statements of Changes in Stockholders’ Equity (Deficiency)
| Common Stock | Additional | Total | ||||||||||||||||||
| Number of | paid-in | Accumulated | Stockholders’ equity | |||||||||||||||||
| Shares | Amount | Capital | deficit | (deficiency) | ||||||||||||||||
| U.S dollars (in thousands) | ||||||||||||||||||||
| For the three-month period ended March 31, 2024 | ||||||||||||||||||||
| Balance as of January 1, 2024 | ( | ) | ||||||||||||||||||
| - | - | |||||||||||||||||||
| Stock-based compensation | ||||||||||||||||||||
| Loss for the period | - | ( | ) | ( | ) | |||||||||||||||
| Balance as of March 31, 2024 | ( | ) | ( | ) | ||||||||||||||||
| Common Stock | Additional | Total | ||||||||||||||||||
| Number of | paid-in | Accumulated | Stockholders’ | |||||||||||||||||
| Shares | Amount | Capital | deficit | equity | ||||||||||||||||
| U.S dollars (in thousands) | ||||||||||||||||||||
For the three-month period ended March 31, 2023 | ||||||||||||||||||||
| Balance as of January 1, 2023 | ( | ) | ||||||||||||||||||
| Stock-based compensation | * | |||||||||||||||||||
| Loss for the period | - | ( | ) | ( | ) | |||||||||||||||
| Balance as of March 31, 2023 | ( | ) | ||||||||||||||||||
| (*) |
The accompanying notes are an integral part of these interim unaudited condensed consolidated financial statements.
| F-37 |
IR-Med Inc.
Interim Unaudited Condensed Consolidated Statements of Cash Flows
| For the three-month period ended | ||||||||
| March 31 | March 31 | |||||||
| 2024 | 2023 | |||||||
| U.S dollars (in thousands) | ||||||||
| Cash flows from operating activities | ||||||||
| Loss for the period | ( | ) | ( | ) | ||||
| Adjustments to reconcile loss for the period to net cash used in operating activities: | ||||||||
| Stock based compensation | ||||||||
| Depreciation | ||||||||
| Accrued financial expenses (income) | ( | ) | ||||||
| Decrease (increase) in accounts receivable | ( | ) | ||||||
| Increase (decrease) in trade and other payables | ( | ) | ||||||
| Net cash used in operating activities | ( | ) | ( | ) | ||||
| Effect of exchange rate changes on cash and cash equivalents | ( | ) | ||||||
| Net decrease in cash and cash equivalents | ( | ) | ( | ) | ||||
| Cash and cash equivalents as at the beginning of the period | ||||||||
| Cash and cash equivalents as at the end of the period | ||||||||
The accompanying notes are an integral part of these interim unaudited condensed consolidated financial statements.
| F-38 |
IR-Med Inc.
Notes to the Interim Unaudited Condensed Consolidated Financial Statements
Note 1 - General
| A. | Description of Business |
IR-Med, Inc. (OTC QB: IRME, hereinafter: the “Parent Company”) was incorporated in Nevada in 2007. IR-Med, Inc. was previously named International Display Advertising, Inc. and changed its name to IR-Med, Inc. in January 2021.
The registered office of IR-Med, Inc. and the corporate headquarters and research facility of IR. Med, Ltd. are located in Rosh Pina, Israel. The Parent Company and IR. Med Ltd. (Hereinafter: the “Subsidiary”) are at times collectively referred to as the “Company”.
On April 9, 2024, the Company’s first device, the PressureSafe, decision support device received a U.S. Food and Drug Administration (FDA) listing certification. PressureSafe is classified as a Class I device. Following the listing certification of the PressureSafe device, the Company has started the preparations for the commercial launch of its first device, the PressureSafe. The Company is developing its technology through its Subsidiary and is utilizing Infra-Red-light spectroscopy (IR) combined with an Artificial Intelligence (AI) technology platform to develop non-invasive devices for various medical indications, by detecting and measuring various biomarkers and molecules in the blood and in human tissue in real-time. The initial product candidates which are currently in various stages of development are non-invasive, user friendly and designed to address the medical needs of large and growing target patient groups by offering earlier and more accurate information for detection, which is expected to reduce healthcare expenses and reduce the widespread reliance on antibiotics administration, and other interventional options optimizing the delivery of targeted medical services.
On January 25, 2024, the Israel
Innovation Authority (the “IIA”) approved the Company’s proposed program to develop a device for the early detection
of diabetic foot ulcers among diabetic patients, with a project budget of NIS
| B. | Going Concern |
The
Company has started the preparations of the commercial launch of its first device, the PressureSafe, but does not expect to generate
significant revenue until such time as the Company shall have completed the design and development of its initial products candidates
and initiates marketing activities for its commercial product. During the three months ended March 31, 2024, the Company incurred losses
of $
Management’s plans regarding these matters include continued development and marketing of the Company’s products, as well as seeking additional financing arrangements. Although management continues to pursue these plans, there is no assurance that the Company will be successful in raising the needed capital from revenues or financing on commercially acceptable terms. The financial statements do not include any adjustments that might result from the outcome of this uncertainty. Management’s plans regarding these matters include continued development and marketing of its products, as well as seeking additional financing arrangements. As a result of the Company’s financial condition substantial doubt exists that the Company will be able to continue as a going concern for one year from the issuance date of this first quarter of 2024 Report.
Following the brutal attacks on Israel, the mobilization of army reserves and the Israeli Government declaring a state of war (the “Iron Swords War”) in October 2023, there has been a decrease in Israel’s economic and business activity. The security situation has led, inter alia, to a disruption in the chain of supply and production, a decrease in the volume of national transportation, a shortage in manpower as well as a decrease in the value of financial assets and a rise in the exchange rate of foreign currencies in relation to the shekel. At this time, the Company has assessed, on the basis of the information it has as of the date of the approval of these financial statements, that the current events and the escalation in security in Israel, may have a material effect on the business plans of the Company in the short term. As a result of the movement and work restrictions in Israel, the Company has begun operating on a limited scale. These restrictions and the shortage in manpower may cause delays in the Company’s research and development activities and in its marketing efforts. In addition, the situation has brought further difficulties in management’s efforts to seek additional financing arrangements. Since this is an event that is not under the control of the Company and matters such as the fighting continuing or stopping may affect the Company’s assessments, as of the reporting date the Company is unable to assess the extent of the effect of the Iron Swords War on its business.
Note 2 - Interim Unaudited Financial Information
The accompanying unaudited financial statements of the Company have been prepared in accordance with U.S. generally accepted accounting principles (“U.S GAAP”) for interim financial information. Accordingly, they do not include all of the information and footnotes required by U.S. GAAP for complete financial statements and therefore should be read in conjunction with the Company’s Annual Report on for the year ended December 31, 2023.
| F-39 |
IR-Med Inc.
Notes to the Interim Unaudited Condensed Consolidated Financial Statements
Note 2 - Interim Unaudited Financial Information (Cont’d)
In the opinion of management, all adjustments considered necessary for a fair statement, consisting of normal recurring adjustments, have been included. Operating results for the three months period ended March 31, 2024 and 2023 and cash flow for the three months period ended March 31, 2024 and 2023 are not necessarily indicative of the results that may be expected for the year ending December 31, 2024.
Use of Estimates:
The preparation of financial statements in conformity with U.S GAAP requires management to make judgments, estimates and assumptions that affect the reported amounts of assets and liabilities and disclosures of contingent assets and liabilities at the date of the consolidated financial statements, and the reported amounts of expenses during the reporting period. Significant items subject to such estimates and assumptions including fair value of share-based compensation and legal claims. Actual results could differ from those estimates.
Note 3 - Significant Accounting Policies
These interim unaudited condensed consolidated financial statements have been prepared according to the same accounting policies as those discussed in the Company’s Annual Report for the year ended December 31, 2023.
Note 4 - Stockholders’ Loans
On March 1, 2024, the Company and the lenders agreed to extend the repayment date to December 31, 2025.
Shareholders’ loans with terms that were amended after the reporting date are considered in determining the classification of debt at the reporting date. Due to agreements reached in 2024 between the shareholders and the Company regarding the repayment date of the loan, the shareholders’ loans on March 31, 2024, are classified as non-current liabilities.
On December 23, 2020 the Company’s board of directors approved and the shareholders adopted a share-based compensation plan (“2020 Incentive Stock Plan”) for future grants by the Company to officers, directors, employees and consultants.
As of March 31, 2024, the Company awarded to its employees and service providers options to purchase up to shares of Common Stock, of which options for shares were at an exercise price of $ per share, options for shares were at an exercise price of $ per share, options for shares were at an exercise price of $ per share. As of March 31, 2024 options for shares were vested with a weighted average of exercise of $ and the remaining balance has a vesting period ranging between to . The options are exercisable for periods ranging between to from the vesting date.
| Weighted average of exercise price | Number of options | |||||||
| Outstanding as of beginning of year | $ | |||||||
| Cancelled | $ | ( | ) | |||||
| Outstanding as of March 31 ,2024 | $ | |||||||
The aforementioned grants were approved following the adoption of the 2020 incentive stock plan and the adoption of the sub plan (the “Israeli appendix”) on April 29, 2021. The Company recorded in the statement of operations a non-cash expense of $ thousand and $ thousand during the three months ended March 31, 2024 and 2023 respectively.
| For the three-month period ended | ||||||||
| March 31, 2024 | March 31, 2023 | |||||||
| US Dollars (In thousands) | ||||||||
| Research and development expenses | ||||||||
| Marketing expenses | ||||||||
| General and administrative expenses | ||||||||
| F-40 |
IR-Med Inc.
Notes to the Interim Unaudited Condensed Consolidated Financial Statements
Note 5 - Stock options plan (Cont’d)
| For the three-month period ended | ||||||||
| March 31, 2024 | March 31, 2023 | |||||||
| Dividend yields (see (I) below) | % | % | ||||||
| Share price (in U.S. dollar) (see (II) below) | , | |||||||
| Expected volatility (see (III) below) | % - | % | % - | % | ||||
| Risk-free interest rates (see (IV) below) | % - | % | % - | % | ||||
| Expected life (in years) (see (V) below) | - | - | ||||||
| I. | ||
| II. | ||
| III. | ||
| IV. | ||
| V. |
Note 6 - Contingent Liabilities and Commitments
On
May 29, 2023, a lawsuit was filed against the Company, the Subsidiary and Mr. Aharon Klein (the “Plaintiff”), a Company Director
and the Company’s Chief Technology Officer in the Tel Aviv District Court of Israel, by an individual who provided, on part time
basis, certain consulting services to the Subsidiary between October 2015 and October 2016, before the acquisition of the Subsidiary
by the Company. The suit alleges breach of contract by the defendants based on non-payment of amounts purportedly owed to the Plaintiff
in respect of the services rendered, including the market value of the Company’s common stock that the Plaintiff alleges should
have been issued to him in respect of services. The suit seeks declaratory judgment that the defendants breached certain agreements with
the Plaintiff and claimed damages in the aggregate amount of approximately $
The Company records a provision in its financial statements to the extent that it concludes that a contingent liability is probable, and the amount thereof is reasonably estimable. Based upon the status of the case described above, management’s assessments of the likelihood of damages, and the advice of counsel, no provisions have been made regarding the matter disclosed in this note. Litigation outcomes and contingencies are unpredictable, and excessive verdicts can occur. Accordingly, management’s assessments involve complex judgments about future events and often rely heavily on estimates and assumptions.
| F-41 |

_______ Units
Each Unit Consisting of
One Share of Common Stock
And
One Warrant to Purchase One Share of Common Stock
PROSPECTUS
Sole Book-Running Manager
Maxim Group LLC
, 2024
Until , 2024 (25 days after the date of this prospectus), all dealers that buy, sell or trade our common stock, whether or not participating in this offering, may be required to deliver a prospectus. This is in addition to the dealers’ obligation to deliver a prospectus when acting as underwriters and with respect to their unsold allotments or subscriptions.
PART II
INFORMATION NOT REQUIRED IN PROSPECTUS
ITEM 13. OTHER EXPENSES OF ISSUANCE AND DISTRIBUTION
The following table sets forth the costs and expenses, other than the underwriting discounts and commissions, payable by IR-Med, Inc., or the Registrant, in connection with the sale of our common stock being registered. All amounts are estimates except for the Securities and Exchange Commission, or SEC, registration fee, the Financial Industry Regulatory Authority, or FINRA, filing fee and the Nasdaq Capital Market, or Nasdaq, listing fee.
| SEC registration fee | $ | 2,439.54 | ||
| FINRA filing fee | $ | 2,979.20 | ||
| Initial Nasdaq Capital Market listing fee | $ | 5,000.00 | ||
| Accounting fees and expenses | $ | 5,000.00 | ||
| Legal fees and expenses | $ | 200,000.00 | ||
| Miscellaneous fees and expenses | $ | 5,000.00 | ||
| Total | $ | 215,418.74.00 |
ITEM 14. INDEMNIFICATION OF OFFICERS AND DIRECTORS
Our Amended and Restated Articles of Incorporation and our Amended and Restated Bylaws provide that each person who was or is made a party or is threatened to be made a party to or is otherwise involved (including, without limitation, as a witness) in any action, suit or proceeding, whether civil, criminal, administrative or investigative, by reason of the fact that he or she is or was one of our directors or officers or is or was serving at our request as a director, officer, employee or agent of another corporation, partnership, joint venture, trust or other enterprise, whether the basis of such action, suit or proceeding is alleged action in an official capacity as a director, officer or trustee or in any other capacity while serving as a director, officer or trustee, shall be indemnified and held harmless by us to the fullest extent authorized by the Nevada Revised Statutes, or NRS, against all expense, liability and loss (including attorneys’ fees and amounts paid in settlement) reasonably incurred or suffered by such.
NRS 78.7502 permits a corporation to indemnify any director or officer of the corporation against expenses (including attorneys’ fees) and amounts paid in settlement actually and reasonably incurred in connection with any action, suit or proceeding brought by reason of the fact that such person is or was a director or officer of the corporation, if such person (i) is not liable pursuant to NRS 78.138 and (ii) acted in good faith and in a manner which he or she reasonably believed to be in or not opposed to the best interests of the corporation, and, with respect to any criminal action or proceeding, had no reasonable cause to believe the conduct was unlawful. In a derivative action (i.e., one brought by or on behalf of the corporation), indemnification may be provided only for expenses actually and reasonably incurred by any director or officer in connection with the defense or settlement of such an action or the suit if such person (i) is not liable pursuant to NRS 78.138 and (ii) acted in good faith and in a manner which he or she reasonably believed to be in or not opposed to the best interests of the corporation, except that no indemnification shall be provided if such person shall have been adjudged to be liable to the corporation, unless and only to the extent that the court in which the action or suit was brought or some other court of competent jurisdiction determines that such person is fairly and reasonably entitled to indemnity for such expenses as the court deems proper.
Our Amended and Restated Articles of Incorporation provide that the liability of our directors and officers shall be eliminated or limited to the fullest extent permitted by the NRS. NRS 78.138(7) provides that, subject to limited statutory exceptions and unless the articles of incorporation or an amendment thereto (in each case filed on or after October 1, 2003) provide for greater individual liability, a director or officer is not individually liable to a corporation or its stockholders or creditors for any damages as a result of any act or failure to act in his or her capacity as a director or officer unless it is proven that: (i) the act or failure to act constituted a breach of his or her fiduciary duties as a director or officer, and (ii) the breach of those duties involved intentional misconduct, fraud or a knowing violation of law.
| II-1 |
ITEM 15. RECENT SALES OF UNREGISTERED SECURITIES
The following summarizes all sales of unregistered securities by us within the past three years:
Between December 24, 2020 and April 20, 2021, we issued to certain accredited investors an aggregate of 9,110,938 units of our securities, with each Unit comprised of (i) two (2) shares of our common stock and (ii) one common stock purchase warrant to purchase an additional share of our common stock at a per share exercise price of $0.64, for aggregate gross proceeds to us of approximately $5.83 million. After deducting for placement related expenses, the aggregate net proceeds from the Private Placement were approximately $5.53 million. The issuance and sale of all such shares has not been registered under the Securities Act, and such shares were issued and sold in reliance upon an exemption from registration afforded by Section 4(a)(2) of the Securities Act and Rule 506 of Regulation D promulgated under the Securities Act as a transaction by an issuer not involving any public offering.
Between April 2022 through July 2022, the Company entered into a securities purchase agreement with six accredited investors providing for the issuance and sale to such investors of an aggregate of 4,119,321 shares of the Company Common Stock and warrants for an additional 4,119,321 shares of the Company Common Stock, exercisable through 2024, at a per share exercise price of $1.10. subject to certain limited adjustments. The aggregate gross proceeds from the private placement were approximately $3,625,000.
On June 12, 2023, we entered into a subscription agreement with one investor pursuant to which we issued 1,000,000 shares of its common stock at a per share price of $1.00 and warrants to purchase up to an additional 1,000,000 shares of common stock at a per share exercise price of $1.40 and expire on the third anniversary from the date of issuance of the warrant to the holder. We are entitled to accelerate the warrant exercise period for all or a part of the then outstanding warrants by written notice to the holders if the publicly traded price of our common stock equals or exceeds $2.50 per share (which amount may be adjusted for certain capital events, such as stock splits, as described herein) and the corresponding average daily trading volume during such period shall equal or exceed 75,000 shares, in each case for the preceding forty (40) consecutive trading days. We received aggregate gross proceeds of $1,000,000 from this financing.
ITEM 16. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
| II-2 |
| * |
To be filed by amendment |
| ** | Filed herewith |
@ Management Contract or Compensatory Plan Arrangement
| II-3 |
(b) Financial Statement Schedules.
Schedules not listed above have been omitted because the information required to be set forth therein is not applicable or is shown in the consolidated financial statements or notes thereto.
ITEM 17. UNDERTAKINGS
The undersigned registrant hereby undertakes:
(1) To file, during any period in which offers or sales are being made, a post-effective amendment to this registration statement:
(i) To include any prospectus required by Section 10(a)(3) of the Securities Act of 1933;
(ii) To reflect in the prospectus any facts or events arising after the effective date of the registration statement (or the most recent post-effective amendment thereof) which, individually or in the aggregate, represent a fundamental change in the information set forth in the registration statement. Notwithstanding the foregoing, any increase or decrease in volume of securities offered (if the total dollar value of securities offered would not exceed that which was registered) and any deviation from the low or high end of the estimated maximum offering range may be reflected in the form of prospectus filed with the Securities and Exchange Commission pursuant to Rule 424(b) if, in the aggregate, the changes in volume and price represent no more than a 20% change in the maximum aggregate offering price set forth in the “Calculation of Registration Fee” table in the effective registration statement; and
(iii) To include any material information with respect to the plan of distribution not previously disclosed in the registration statement or any material change to such information in the registration statement;
(2) That, for the purpose of determining any liability under the Securities Act of 1933, each such post-effective amendment shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
(3) To remove from registration by means of a post-effective amendment any of the securities being registered which remain unsold at the termination of the offering.
(4) That, for the purpose of determining liability under the Securities Act of 1933 to any purchaser, each prospectus filed pursuant to Rule 424(b) as part of a registration statement relating to an offering, other than registration statements relying on Rule 430B or other than prospectuses filed in reliance on Rule 430A (§230.430A of this chapter), shall be deemed to be part of and included in the registration statement as of the date it is first used after effectiveness. Provided, however, that no statement made in a registration statement or prospectus that is part of the registration statement or made in a document incorporated or deemed incorporated by reference into the registration statement or prospectus that is part of the registration statement will, as to a purchaser with a time of contract of sale prior to such first use, supersede or modify any statement that was made in the registration statement or prospectus that was part of the registration statement or made in any such document immediately prior to such date of first use.
Insofar as indemnification for liabilities arising under the Securities Act of 1933 may be permitted to directors, officers and controlling persons of the registrant pursuant to the foregoing provisions, or otherwise, the registrant has been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Securities Act of 1933 and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Securities Act of 1933 and will be governed by the final adjudication of such issue.
| II-4 |
SIGNATURES
Pursuant to the requirements of the Securities Act of 1933, as amended, the Registrant certifies that it has reasonable grounds to believe that it meets all of the requirements for filing on Form S-1 and has duly caused this Registration Statement to be signed on its behalf by the undersigned, thereunto duly authorized, on this 24 day of June 2024.
| IR-MED, INC. | ||
| By: | /s/ Aharon Klein | |
| Aharon Klein | ||
| Interim Chief Executive Officer (Principal Executive Officer) | ||
POWER OF ATTORNEY
KNOW ALL BY THESE PRESENTS, that each person whose signature appears below hereby constitutes and appoints Aharon Klein and Sharon Levkoviz, and each of them, his true and lawful agent, proxy and attorney-in-fact, with full power of substitution and resubstituting, for him and in his name, place and stead, in any and all capacities, to (i) act on, sign and file with the Securities and Exchange Commission any and all amendments (including post-effective amendments) to this registration statement together with all schedules and exhibits thereto and any subsequent registration statement filed pursuant to Rule 462(b) under the Securities Act of 1933, as amended, together with all schedules and exhibits thereto, (ii) act on, sign and file such certificates, instruments, agreements and other documents as may be necessary or appropriate in connection therewith, (iii) act on and file any supplement to any prospectus included in this registration statement or any such amendment or any subsequent registration statement filed pursuant to Rule 462(b) under the Securities Act of 1933, as amended and (iv) take any and all actions which may be necessary or appropriate to be done, as fully for all intents and purposes as he might or could do in person, hereby approving, ratifying and confirming all that such agent, proxy and attorney-in-fact or any of his substitutes may lawfully do or cause to be done by virtue thereof.
Pursuant to the requirements of the Securities Act, this Registration Statement has been signed by the following persons in the capacities and on the dates indicated.
| Signature | Title | Date | ||
| /s/ Oded Bashan | Chairman of the Board | June 24, 2024 | ||
| Oded Bashan | ||||
| /s/ Aharon Klein | Chief Executive Officer, Chief Technology Officer, Director | June 24, 2024 | ||
| Aharon Klein | (Principal Executive Officer) | |||
| /s/ Sharon Levkoviz | Chief Financial Officer | June 24, 2024 | ||
| Sharon Levkoviz | (Principal Financial and Accounting Officer) | |||
| /s/ Ohad Bashan | Director | June 24, 2024 | ||
| Ohad Bashan | ||||
| /s/ Ron Mayron | Director | June 24, 2024 | ||
| Ron Mayron | ||||
| /s/ Yaniv Cohen | Director | June 24, 2024 | ||
| Yaniv Cohen | ||||
| /s/ Avital Rosenberg | Director | June 24, 2024 | ||
| Avital Rosenberg |
| II-5 |